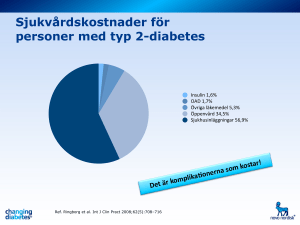
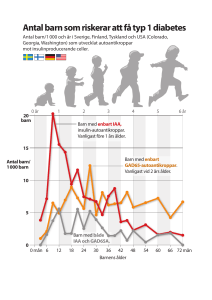
Molekylerna bakom diabetes
advertisement

Molekylerna bakom diabetes Helena Edlund professor, utvecklingsbiologi, Umeå centrum för molekylär medicin, UCMM Färgversioner av bilderna till detta föredrag kan ses på www.medfak.umu.se/forskning/forskningens-dag/-forskningens-dag-2012/ Vid Forskningens dag i Piteå konstaterade vi att jag är ensam om att inte ha frågetecken efter rubriken i programmet, men jag vill gärna lägga till undertiteln ”Insulin på gott och ont”? Det är helt klart att vi behöver insulin, men större delen av mitt föredrag kommer att visa att man kan få för mycket av det goda och att det kan bli en del av problemet, framför allt när det gäller typ 2-diabetes. I min forskargrupp vid UCMM har vi under lång tid använt musen som modellsystem för att förstå, till att börja med, hur bukspottkörteln bildas, se Figur 1. Utvecklingen går från ett tidigt anlag med två knoppar, som man kan kalla bukspottkörtelstamceller, fram till det fullt utvecklade organ som ni kunde se så fint på de tredimensionella bilder Nina Fransén Pettersson visade. Huvuddelen av bukspottkörteln, 95 procent av volymen, producerar och transporterar matsmältningsenzymer som utsöndras till tarmen och hjälper till att bryta ner maten. Den delen kallas exokrina pankreas och innehåller i huvudsak två celltyper: acinära celler, som producerar matspjälkande enzymer, och gångceller, som transporterar enzymerna. Vår forskning är främst inriktad på endokrina pankreas, som består av små ansamlingar av hormonproducerande celler, de langerhanska öarna, se Figur 2. I hela bukspottkörteln finns de här cellansamlingarna som faktiskt innehål- 70 Figur 1. Tidig utveckling av bukspottkörteln (pankreas) hos mus. De vänstra bilderna visar läget efter ca 4 veckor hos mus och ca 9 veckor hos människa, mittbilderna 4,5 respektive 10 veckor, de högra bilderna 5 respektive 12 veckor. Figur 2. I bukspottkörteln produceras insulin och flera andra hormoner av betaceller i de s.k. langerhanska cellöarna – en sådan ö visas i förstoring till höger. 71 ler fem olika typer av hormonproducerande celler: a-celler (alfaceller, 15–20 % av totalmängden) som tillverkar glukagon; b-celler (betaceller, 60–80 %) med insulin; d-celler (deltaceller, 5–10 %) med somatostatin; PP-celler (mindre än 2 %) med pankreatisk polypeptid samt e-celler (epsilonceller, mindre än 1 %) med ghrelin. Naturligtvis har vi främst varit intresserade av att förstå de insulinproducerade betacellerna, de som är infärgade i vävnadssnittet på den förstorade delen av Figur 2. Vårt övergripande mål är att försöka identifiera de molekylära mekanismerna bakom utvecklingen av diabetes, och det gör vi genom att studera de gener och mekanismer som styr bildningen av betacellerna och som säkerställer deras funktion. Hur får vi fungerande betaceller och hur bibehåller vi deras funktion i den vuxna körteln? Som ni redan har hört idag är överlevnaden hos bukspottkörtelcellerna ett problem vid typ 1-diabetes, då betacellerna förstörs av kroppens egna immunförsvarsceller. Det finns också, som jag kommer att nämna helt kort, studier som tyder på att vi tappar insulinproducerande celler även vid utveckling av typ 2-diabetes. Behandlingsförsök med stamceller Vid typ 1-diabetes förstör kroppen eget immunförsvar de insulinproducerande cellerna. Det finns en behandling, att ta insulin, som är livslång och egentligen ingen bot. Som Nina Fransén Pettersson nämnde finns det emellertid en metod som man skulle kunna kalla bot mot sjukdomen: Att transplanera in nya cellöar. Problemet där är framför allt tillgången på material som kan doneras. Liksom vid alla andra former av organtransplantation råder en brist på donationsmaterial och i det här fallet krävs det ofta två donatorer till varje patient, vilket betyder att det knappast finns eller kommer att finnas tillräckligt med cellöar att tranplantera. Vår forskning skulle möjligen kunna bli till nytta vid typ 1-diabetes genom att öppna vägen för en stamcellsbaserad behandling, det vill säga att få olika typer av omogna celler i kroppen att utvecklas till betaceller. Det skulle göra det möjligt att få ganska stor tillgång till insulinproducerande celler. Stamceller som sådana tillverkar emellertid inte insulin och frågan är hur vi ska nå dit. Vilka typer ska vi utgå från: Embryonala stamceller (från embryon), fetala stamceller (från foster), vuxna stamceller, kroppsegna stamceller? Oavsett vilken typ vi väljer att starta med måste vi veta hur vi 72 får den att utvecklas till en insulinproducerande betacell. Det är just den frågan vi har ägnat oss åt genom att studera det normala förloppet under fosterutvecklingen. Vilka gener och vilka faktorer styr egentligen att de omogna cellerna blir just insulinproducerande celler? En annan sak vi måste vara säkra på att hur vi får de celler som vi transplanterar – om de nu är utvecklade utanför kroppen eller donerade cellöar – att överleva. Med andra ord: Hur får vi dem att tolereras av kroppen så att de inte stöts bort? Även om vi löser det problemet genom att använda kroppsegna stamceller kvarstår problemet med immunförsvarets angrepp. Inte minst gäller det att kunna säkerställa de nya cellernas funktioner och överlevnad över lite längre tid. Med dagens transplantationer av cellöar kan man få väldigt positiva effekter. Nina gav ett exempel på en patient som klarat sig utan att ta insulin i 13 år, men oftast måste man fylla på med nya cellöar efter bara några år, kanske 5 år eller tidigare. Figur 3. I olika skeden av utvecklingen från befruktning (överst) via embryo fram till färdig bukspottkörtel finns möjligheter (markerade med pilar) att isolera stamceller och betaceller för vidare utveckling och/eller transplantation vid typ 1-diabetes. 73 Fosterutvecklingen utgår ju från det befruktade ägget, ”urstamcellen”, men på vägen till att bli betaceller, nedåt i Figur 3, ställs cellerna hela tiden inför olika val. Omgivningen och olika faktorer från de angränsande cellerna ”lotsar” dem fram till att faktiskt bli insulinproducerande. Det sker hela tiden under fosterutvecklingen. Om vi till exempel hoppar fram till de celler som ”vet” att de ska bli en del av bukspottkörteln, vet de ännu inte vilket typ av cell de ska bli, exokrin eller endokrin. När de väl vet att de ska bli endokrina, alltså hormonproducerande, finns det olika vägar att ta där också. Som forskare måste vi kunna återskapa det här förloppet genom att lotsa cellerna på exakt rätt sätt hela vägen ner till att bli fungerande betaceller. När vi försöker efterlikna det här kan vi idag faktiskt ta mänskliga embryonala stamceller (hES på den högra bildserien i Figur 3) – inte riktigt det befruktade ägget utan några steg senare – och isolera dem i en odlingsskål. De här kan åtminstone teoretiskt utvecklas till alla celler i kroppen. Vi kan också få dem att ta ett utvecklingssteg till så att de blir endodermala och vi kan få dem att bilda tidiga bukspottkörtelstamceller, enligt bildserien till höger i figuren. Frågan är då hur vi ska få dem att gå hela vägen till att bli insulinproducerande betaceller. Idag kan vi, och flera andra forskargrupper, faktiskt få fram insulinproducerande celler med utgångspunkt från embryonala stamceller, men vi får bara ut ett fåtal, och den stora knäckfrågan just nu är hur vi kan styra de här tidiga cellerna: Hur får vi dem att gå hela vägen och hur kan vi få ut tillräckligt många? Där står vi idag. Vi kan nå fram till bukspottkörtelstamceller, men vi har inte knäckt utmaningen att komma hela vägen till fullt ut fungerande betaceller. Figur 4 sammanfattar en studie som publicerades för några år sedan av amerikanska forskare vid ett bioteknikföretag i Kalifornien. De hoppade över det sista steget utanför kroppen och transplanterade bukspottkörtelstamcellerna direkt till musen. Efter några månader fick de en utmognad av hormonproducerande celler i bukspottkörteln, också insulinproducerande celler. De här mognade stamcellerna kunde faktiskt bidra till musens blodsockerreglerande förmåga. Det vi vill åstadkomma är att kunna göra det här helt och hållet utanför kroppen, eftersom man i annat fall skulle transplantera in omogna stamceller i musens kropp. Även om man då skulle kunna få insulinproducerande celler att bildas uppstår andra problem längs vä74 Figur 4. I en studie från USA har man lyckats utveckla bukspottkörtelstamceller från humana embryonala stamceller som sedan kan mogna och bilda insulinproducerande celler i musens kropp, dvs. in vivo. Jfr. Figur 3. (Kroon et al. Nat. Bio. 2008) gen, framför allt tumörbildning. Det är skälet till att vi måste hitta ett sätt att styra alla celler till att bli fullt utmognade betaceller. Vi måste kunna utesluta att någon enda stamcell inte har hunnit mogna när den transplanteras, därför att då riskerar vi tumörer. Vi måste också säkerställa att de betaceller vi lotsar fram från stamcellerna svarar normalt på blodsocker och utsöndrar insulin. Ett problem med de insulinproducerande celler som man hittills har kunnat ta fram utanför kroppen är att de, även om de kan producera insulin, inte svarar på blodsockret och därför inte utsöndrar insulin. Än vet vi emellertid inte om det räcker med att ta fram betaceller. Måste vi återskapa en hel langerhansk ö där alla de andra hormonproducerande cellerna finns med? En annan fråga är var ska vi transplantera in cellerna. Levern är inte det bästa eftersom det är en miljö där ganska många giftämnen ansamlas; vi talar om kroppens avgiftningscentral. Det är en del av förklaringen till att de transplanterade öarna inte har klarat sig så bra där. Kanske kan man transplantera cellerna till ögat som Nina Fransén Pettersson visade? Det pågår sådana försök på apor, inte bara för att kunna studera betaceller utan också för att försöka bota diabetes. Det har dessutom gjorts försök att stoppa in cellerna i muskler, till exempel i överarmen. 75 En okänslighet mot insulin Nu går jag över till att diskutera typ 2-diabetes och vår forskning om den sjukdomen. Som vi har fått veta yttrar sig typ 2-diabetes som brist på verksamt insulin. Kroppen har en utvecklat en okänslighet för insulinet, insulinresistens. Det finns dessutom tecken på att kroppen tappar betaceller. Den som har gått länge med typ 2-diabetes har ett lägre antal betaceller. Det betyder att också här kan man få en avdödning av de insulinproducerande cellerna. När man har försökt hitta arvsanlag (gener) som ökar risken för diabetes ser man att de flesta som har identifierats är kopplade till funktionen hos betacellerna, som i Figur 5, medan man inte har funnit någon gen som är kopplad till insulinokänsligheten. Därför får man nog konstatera att betacellen är central inte bara för typ 1-diabetes utan också för typ 2-diabetes. Figur 5. De flesta identifierade riskgener för typ 2-diabetes är kopplade till minskad funktion hos betacellerna (inringat i listan). Vad gör då insulinet som är bra? Jo, det stimulerar upptaget av socker, glukos, i fettvävnaderna och levern, men framför allt i musklerna som på det sättet förses med energi. Insulinet ska också minska utsöndringen av fria fettsyror från fettvävnaden och levern. Insulinet ska dessutom hämma sockerproduktionen i levern, så att det glykogen som finns lagrat där inte omvandlas och utsöndras i form av socker. Vid insulinokänslighet försvinner eller försämras alla de där funktionerna – och det höjer blodsockernivån. Denna höjning känner de insulinproducerande betacellerna av och försöker kompensera det genom att utsöndra ännu mer insulin för att stimulera 76 Figur 6. Schematisk bild av hur typ 2-diabetes utvecklas. På grund av bland annat fetma utvecklar kroppen en okänslighet för insulin, insulinsresistens, som leder till höga blodsockervärden, hyperglykemi. Den ökade mängden av socker, glukos, i blodet triggar betacellerna i bukspottkörteln att producera ännu mer insulin, vilket ger ett överskott av insulin i kroppen, hyperinsulinemi. vävnaderna att ta upp sockret så att kroppen blir av med det. Men det innebär ju höga insulinnivåer istället, hyperinsulinemi, se Figur 6. Under en stor del av diabetesutvecklingen, den fas som Margareta Norberg kallade pre-diabetes eller förstadium till diabetes, har kroppen normala blodsockervärden men en skyhög insulinnivå. Det är först när betacellen inte längre kan utsöndra insulin effektivt som vi utvecklar typ 2-diabetes, och det är förhållandevis ganska sent, se Figur 7. Det man faktiskt borde fundera på inom vården är att inte bara kontrollera blodsockret utan också insulinnivån i blodet som ett sätt att fånga upp individer som är på väg mot att utveckla diabetes. 77 Figur 7. Det inramade området visar värden under förstadiet till typ 2-diabetes, så kallad pre-diabetes. Typiskt är normalt blodsocker men höga nivåer av insulin. Figur 8. Fettsyrareceptorn Gpr40 på de insulinproducerande betacellerna stimuleras av fettsyror i blodet vilket ökar insulinutsöndringen. Det blå (mörkaste) på bilden till vänster visar receptorn. De övriga bilderna visar att receptorn enbart uttrycks in insulinceller: Insulin visas i rött (mörkgrått) och glukagon i grönt (ljusgrått). 78 Kan man sänka insulinnivåerna? Hur ska man komma åt att bota typ 2-diabetes? Ett sätt är ju att försöka behålla eller återställa betacellernas funktioner. Ett annat att öka insulinkänsligheten så att de vävnader som ska ta upp sockret faktiskt också gör det. Ett tredje att faktiskt försöka sänka insulinnivåerna och om det handlar våra studier av fettsyrereceptorn GPR40. Den sitter på de insulinproducerande betacellerna och känner av fria fettsyror i blodcirkulationen, se Figur 8. Det är fortfarande sockret som stimulerar utsöndringen av insulin, men fettsyrorna förstärker sockrets effekt så att mer insulin utsöndras, ett slags turboeffekt. Vi har också visat att det finns celler i tarmen som känner av fettsyror med den här receptorn, och det leder till att dessa celler i sin tur utsöndrar hormoner som stimulerar betacellerna att utsöndra mer insulin. Receptorn verkar med andra ord både direkt på betacellerna och indirekt via tarmen för att öka insulinutsöndringen vid höga fettsyrenivåer. För att se hur viktig den är för insulinutsöndringen slog vi ut receptorgenen hos möss, och då hände egentligen ingenting: Mössen var helt friska med normal sockertolerans, vilket visar att receptorn inte påverkar den insulinutsöndring som blodsockret stimulerar. Men det handlar ju om en fettsyrereceptor, så vi gick vidare för att se vad som händer om vi satte mössen på en energirik kost för att göra dem feta. När vi då gjorde en sockerbelastning såg vi att de feta kontrollmössen – som hade receptorgenen kvar – var prediabetiska, några hade också en fullt utvecklad diabetes. Däremot var de möss som saknar fettreceptorn GPR40 på betacellerna helt friska och utvecklade inte diabetes. När vi mätte insulinkänsligheten svarade de feta kontrollmössen väldigt dåligt medan de muterade mössen svarade normalt. En stor skillnad låg i nivåerna av insulin: De muterade mössen – utan receptorn – bibehöll normala insulinnivåer och normal insulinutsöndring när man tillför socker. De feta kontrollmössens insulinnivåer blev däremot skyhöga. Insulinöverskott och fettlever Något som kopplar till det Tommy Olsson berättade är att de feta kontrollmössen utvecklade fettlever till skillnad från de muterade mössen. Det är känt sedan en tid att tillståndet med insulinöverskott, hyperinsulinemi, har samband med förstadiet till diabetes men framför allt med utvecklingen 79 Figur 9. Skillnaden i upptaget av fettsyror hos möss med fettsyrareceptorn Grp40 (”kontroll”) och muterade möss utan den (randiga staplar) förklaras med att normalmössen, men inte de muterade mössen, har uppgraderat två gener som stimulerar fett-transporten. HFD = high fat diet (fetmaskapande diet). av fettlever. Vi ville försöka förstå det på molekylär nivå: Var beror skillnaden på? Bildas det mera fett i levern hos kontrollmössen än hos de muterade? Förbränner de muterade mössen mer fett i levern så att de därför inte får fettlever? Eller tar kontrollmössen, som har receptorn, upp mer av fettsyrorna? Vi såg ingen skillnad i hur de bildar och förbränner fett i levern, men de feta mössen med fungerande receptor – våra kontrollmöss – hade uppreglerat två olika gener som stimulerar fettsyreupptag i levern, se Figur 9. Normala möss som blir feta och har höga insulinnivåer tar upp fet80 tet i levern via framför allt den här fett-transportören. Hos våra muterade möss såg vi inte de ökade uttrycken av fettuttagsgenerna. Finns det då något som säger att det skulle fungera på liknande sätt hos människor, att fettupptagsstimulering via den här receptorn ökar risken för diabetes? Som Kristina Lejon berättade kan man ha naturliga variationer i sina gener, och därmed proteiner, som ökar sårbarheten och benägenheten för vissa sjukdomar, men naturligtvis finns också det omvända: variationer som ger ökat skydd och minskar benägenheten att insjukna. Man har funnit en genvariant i Gpr40-receptorn hos människan där man har en aminosyra, en byggsten i proteinet, på en viss position i genen, His211 på fackspråk. Det för med sig att receptorn svarar så dåligt att den i princip inte fungerar: De insulinproducerande cellerna ser då inte och reagerar inte på fettsyrorna. Personer med den genvarianten har en lägre benägenhet för typ 2-diabetes vid varje givet BMI-värde. Det antyder att om vi inte har den här receptorn, eller om den som i det här fallet inte fungerar riktigt bra, utvecklar vi inte diabetes eller löper i varje fall inte samma risk. Om man ska sammanfatta vet vi att alltså insulinresistens är en faktor bakom typ 2-diabetes. Till följd av resistensen höjs blodsockernivåerna, vilket betacellerna försöker kompensera med att utsöndra mer insulin. Samtidigt visar våra studier att ökade nivåer av fettsyror, via GPR40-receptorn, i sig ger ökade insulinnivåer. Insulinet blir då en del av problemet eftersom det driver på inlagringen av fett i levern med insulinresistens i levern som följd. Därmed får vi en ond cirkel som driver diabetesutvecklingen framåt, se Figur 10. Det vi försöker titta på är hur insulinet genom att öka uttrycket av fett-transportören C36 i levern också stimulerar direkt fettsyraupptag och utveckling av fettlever. Vi försöker nu förstå hur det fungerar i detalj genom att använda två musmodeller, en där mössen får dricka vatten med 30 % socker (tre gånger så mycket som i läsk) och en där vi försöker ge insulin direkt till leverceller. Det är pågående studier, men de resultat vi har hittills tyder på att så snart vi får höga insulinnivåer ser vi en uppreglering av den här genen som stimulerar fettsyraupptaget i levern och får ökat fettinnehåll i levern. Fetma och förhöjda blodnivåer av fettsyror får alltså betacellen att utsöndra mer insulin än den skulle gjort om bara socker hade varit närvarande. Det leder till att levern lagrar in fett. Utvecklingen av fettlever är, som 81 Figur 10. När nivåerna av fria fettsyror (FFS) i blodet ökar medför det förhöjda insulinnivåer via GPR40-receptorn på de insulinproducerande betacellerna. Det stimulerar i sin tur upptaget av fett i levern med en insulinokänslighet som följd också där. Den infällda mikroskopbilden visar fettinlagring i levern. Tommy Olsson visade, en viktig komponent som nästan inte går att skilja från diabetesutvecklingen. Sedan är fettlever i sig också ett hälsoproblem. I USA beräknar man att en tredjedel av hela befolkningen har det man kal�lar ”fettrelaterad fettleverutveckling” eller ”icke alkoholrelaterad fettlever”. Det här tillståndet ökar i hela världen, även hos barn där det dessutom går ner i åldrarna: man beräknar att 10 % av alla barn och ungdomar i USA har fettlever. Bland personer med sådan fettlever beräknar man att 60–80 % har insulinresistens. Hos patienter med typ 2-diabetes har 50–60 % fettlever. I sig är fettlever ett tillstånd som kan avhjälpas, precis som Tommy visade, men problemet är att det alltför ofta övergår i en leverinflammation med ärrbildning, vilket kan leda till kroniska leversjukdomar som skrumplever och 82 dessutom öka risken för levercancer. Det här är med andra ord ett stort kliniskt problem som går hand i hand med övervikt, fetma och typ 2-diabetes. Motion för autofagins skull Höga insulinnivåer är alltså inte bra med tanke på fettlever och diabetesutveckling, men det är också en stark riskfaktor för polycystiskt ovariesyndrom (en sjukdom med många små cystor i äggstockarna), för flera cancerformer (framför allt i tarm, bröst och prostata) samt för Alzheimers sjukdom (som ibland brukar kallas typ 3-diabetes). Jag har visat att höga insulinnivåer är ett problem genom att de stimulerar fettinlagring i bland annat levern. En annan viktig aspekt på insulinöverskottet, som vi blivit allt mera intresserade av, är att det faktiskt motverkar en livsviktig process i våra celler som kallas autofagi. Den kan man beskriva som cellens ”städpatrull”: Ett slags membran, hinnor, som fångar in och bryter ner molekyler som på sikt skulle kunna skada cellen om de får bli kvar. Jag brukar tänka på autofagi, eller snarare brist på autofagi, som att när man vänder ryggen till ett städat tonårsrum tar det bara timmar innan det råder fullt kaos och ligger grejor överallt – autofagins uppgift är att städa bort all bråte som i annat fall skulle slamma igen cellen. Autofagin är också en viktig process för att vi ska överleva vid energibrist. Den gör att vi kan klara oss ett tag utan mat, men inte vatten, under en längre tid. För möss är det också visat att det är autofagi som gör att de överlever från födseln fram till dess att de kan börja dia. Om autofagin inte fungerar klara sig inte musungarna så långt. Det handlar alltså om både en städpatrull och en räddningspatrull. Problemet är emellertid att ett överskott på insulin hämmar autofagin via det så kallade TOR-komplexet. För att motverka det här finns en viktigt enzym som heter AMPK (AMP-aktiverat proteinkinas) som också känner av energinivån i cellerna. Det kan aktiveras av en lågkaloridiet eller om energinivåerna går ner av andra skäl, till exempel hos den som tränar. Man vet sedan tidigare att det här enzymet stimulerar upptaget av socker, ökar förbränningen av fett, blockerar bildningen av fettsyror och bromsar bildningen av blodfetter – med andra ord förbättrar det insulinkänsligheten. Därför har läkemedelsindustrin satt stort fokus på APMK som ett sätt att behandla typ 2-diabetes och stimulera insulinkänsligheten. Det kanske mest använda 83 läkemedlet vid typ 2-diabetes just nu, metformin, aktiverar faktiskt AMPK, om än indirekt, och det aktiverar i sin tur autofagin. Nyligen publicerades forskningsrön som visar att om man får möss att motionera aktiveras autofagi i deras muskler och även i andra organ. Det är också visat, i en ganska färsk studie, att de positiva effekterna av motion hos de här mössen på sockerupptaget och därmed insulinkänsligheten i bland annat musklerna har med autofagin att göra. Om mössen fick motionera och hade en defekt i autofagin tappade man de positiva effekterna. Feta möss som fick motionera aktiverade autofagin och tog upp socker i musklerna, men om de feta mössen hade något fel på sin autofagi uteblev hela effekten av motionen. Det verkar alltså vara så att om insulinöverskottet hämmar autofagin kan både motion och en lågkaloridiet stimulera AMPK-enzymet, hämma TOR-komplexet och stimulera autofagin, som i sin tur verkar stimulera AMPK och vi får en ”god cirkel”. För några år sedan kom en studie på apor som visar ungefär samma sak: En grupp apor hade ständig tillgång på mat i sin bur och åt när de ville medan en annan grupp fick samma mat, men portionerad så att kaloriintaget låg 30 % lägre än i den första gruppen. De apor som fick 30% mindre kalorier per dag levde längre: I den fritt ätande gruppen hade hälften dött vid 28 års ålder medan fyra av fem levde i lågkalorigruppen. Dessutom förhindrade man uppkomsten av diabetes helt bland de apor som fick lågkalorikosten medan hälften i den fritt ätande gruppen hade antingen prediabetes eller fullt utvecklad diabetes. Motsvarande lägre risk och insjuknande kunde man se när det gällde hjärt-kärlsjukdomar och cancer. Ett annat exempel är invånarna på den japanska ön Okinawa. De lever längst i hela världen, åtminstone historiskt sett, och har lägre förekomst av sjukdomar som diabetes, hjärt-kärlsjukdom, cancer och höftfrakturer. På Okinawa äter befolkningen ungefär 20 procent färre kalorier än genomsnittsjapanen. Kosten domineras av grönsaker, ris, frukt och sojaprodukter. I allmänhet är portionerna mindre, man äter till ungefär 80 procent av full mättnad. Okinawaborna har regelbunden fysisk aktivitet av typ vardagsmotion snarare än ren träning. Deras genomsnittliga BMI-värde ligger strax över 20. På motsvarande sätt kan man se det som hände i Norge under ockupationen 1940–1945. Där minskade antalet nya fall av typ 2-diabetes fall med 84 Figur 11. Tabellen visar effekterna på typ 2-diabetes av en diet med lågt kaloriintag (600 kcal/dag). Översta raden visar värden för en grupp friska kontrollpersoner (Kontr.), därunder för 11 försökspersoner med typ 2-diabetes (T2D) och nederst för samma personer efter en vecka med lågkaloridieten. Lägg märke till den kraftiga minskningen av insulinvärdena (inramat). 85 procent på grund av matransoneringen, men direkt efter kriget började ökningen igen. Man såg ingen sådan effekt på typ 1-diabetes. Man kan säga att de här rönen kopplar ihop det jag har sagt med det som Margareta Norberg och Tommy Olsson var inne på: Vi kan förebygga livsstilssjukdomar som typ 2-diabetes med kost och motion. Men går det att backa bandet, alltså bli av med sjukdomen? Hösten 2011 kom en studie som jag tycker är en av de mest lovande på länge. Där utgick man från de fetmaoperationer som Margareta nämnde och deras positiva effekt på typ 2-diabetes och flera andra metabola sjukdomar. Effekten kommer snabbt, innan personerna egentligen har gått ner i vikt, och det har funderats om det är tarmfloran som ändras eller kanske hormonbalansen. Men i den här studien utgick forskarna från hypotesen att orsaken kan vara att de här personerna går från ett högt kaloriintag till ett lågt. För att pröva den tanken användes försökspersoner med diagnostiserad typ 2-diabetes som inte hade haft sjukdomen längre än fyra år: 20 personer gick in i studien och 11 fullföljde. De fick inte äta mer än 600 kilokalorier per dag i åtta veckor – och det låter jättetufft, men det positiva är att redan efter en vecka såg man positiva effekter på insulinnivån, som var halverad, på blodsockret och på de fria fettsyrorna (triglycerider), se Figur 11. De här tydliga effekterna kom redan efter en vecka, utan att det egentligen hade hänt så mycket med personernas vikt, genomsnittet hade minskat men BMI låg fortfarande över 30, 85 alltså definitionsmässigt fetma. En stor effekt var just att fettinlagringen i levern sjönk med 30 % på den första veckan. Levern producerade inte längre socker och dess insulinkänslighet var återställd. Det som är viktigt med den här studien är att vi faktiskt har våra betaceller kvar vid typ 2-diabetes och kan få dem att fungera igen. Det är inte kört, åtminstone i den här gruppen av patienter. Det går att göra någonting åt det här tillståndet. 86 Sverker Olofsson: Tillbaka till början av ditt föredrag: Är transplantation framtidens behandling? Helena Edlund: Ja, det tror jag. Om det sedan är stamceller eller något annat är källa till de nya insulinproducerande cellerna, det får vi se, men jag tror inte att vi kommer att kunna täcka behovet av donerade cellöar för att transplantera till patienter med typ 1-diabetes. Vi är inte där än, och jag vet inte om vi någonsin kommer så långt att vi på ett säkert sätt kan få stamceller att gå hela vägen fram till fungerande, insulinproducerande celler och dessutom undvika tumörbildning. Sverker Olofsson: Finns det en forskardröm, eller kanske inte bara en dröm, som säger att det barn som har fått typ 1-diabetes kanske kan komma till rätta med det här med en transplantation? Helena Edlund: Ja, i teorin finns ingenting som säger att det inte skulle fungera. Vi måste bara säkerställa att vi får fram de insulinproducerande cellerna utan risk för tumörbildning, att de fungerar och att de överlever. Om vi kan utveckla den stamcellsbaserade behandlingen får vi många insulinproducerande celler att transplantera. Men hur får vi cellerna att fungera? Var ska vi stoppa dem i kroppen? Hur får vi dem att överleva? De immunförsvarsdämpande medel som ges för att förhindra avstötning påverkar också betacellerna. Det finns alltså mycket kvar att göra, men i slutänden skulle vi med en stamcellsbaserad behandling ha hur mycket celler som helst att transplantera. Sverker Olofsson: Vi som börjar nå mogen ålder, bör vi gå till doktorn och kontrollera insulinnivån? Helena Edlund: Jag tycker det, problemet är att jag tror att det skulle bli för dyrt. Det är viktigt att hålla koll på sitt blodsocker, men också att få reda på sitt insulinvärde. Sverker Olofsson: Och om man har för hög nivå, får man ett piller då? Helena Edlund: Nej, till att börja med räcker det nog med att skära ned på snabba kolhydrater och gå ut och promenera. 87