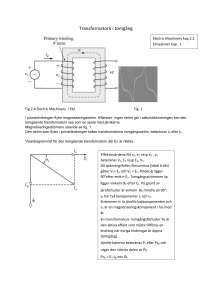
scriptum nr 29 - Forskningsarkivet
advertisement

SCRIPTUM NR 29 Red. Egil Johansson Rapportserie utg. av Forskningsarkivet vid Umeå Universitet ISSN 0284-3161 CODEN: UM/FARK/SC-91/0029 GENEALOGI OCH GENETIK Beskrivning av sex projektområden där kyrkböcker använts i genetisk forskning Stefan Nordström Institutionen för genetik, Umeå univ. FORSKNINGSARKIVET BOX 1441 901 24 UMEÅ Tel. 090/166571 FEBRUARI 1991 Fax. 090/166643 Reprocentralen Samhällsvetarhuset 1991 Redaktörens rader Forskningsarkivet i Umeå syftar till en nära samverkan mellan arkiven och forskningen vid universiteten. Fördenskull utger Forskningsarkivet källskrifter i skriftserien URKUNDEN. Där publiceras valda akter och dokument ur våra arkiv, som blivit aktuella i pågående forskning och utbildning vid universitetet. På motsvarande sätt presenteras vetenskapliga framställningar och bearbetningar av det historiska källmaterialet i rapportserien SCRIPTUM. Syftet med denna serie kan anges i följande huvudpunkter. Publikationsserien SCRITPUM skall 1. utge forskningsmässiga kommentarer till utgåvor av källskrifter i Forskningsarkivets källserie URKUNDEN, 2. publicera andra forskningsrapporter med anknytning till Forskningsarkivets verksamhet, som befinns vara angelägna för den vetenskapliga metodutvecklingen och debatten, 3. publicera framställningar av till exempel lokalhistorisk karaktär av mer allmänt intresse för Forskningsarkivets verksamhet och för en bredare allmänhet. Härmed önskar Forskningsarkivet inbjuda alla intresserade till läsning och till att genom egna bidrag deltaga i utgivningen av skriftserien SCRIPTUM för ett ökat meningsutbyte inom och mellan olika discipliner vid vårt och andra lärosäten. För Forskningsarkivet i Umeå Egil Johansson Tillägnas min egen F2-generation, särskilt yngsta barnbarnet MIKAEL, född den 6 december 1990. P = parental(föräldra)generationen F j = första filialgenerationen F2 = andra filialgenerationen 1 INNEHÅLLSFÖRTECKNING Sid. Inledning 3 Genetik och genetisk genealogi Livets gåta Spekulationer och forskning kring ärftlighet Genetik - Genesis Genetisk genealogi 5 5 5 7 8 Hereditär maculadegeneration HMD och husförhör Genkälla i Vilhelmina, Västerbottens län Primär genkälla i Älvdalen, Kopparbergs län Genen har "utvandrat" till USA Varierande symtombild Genbärare spåras med EOG Homozygota anlagsbärare Varierande debutålder Försök att isolera och molekylärt definiera genen för HMD 11 11 15 17 20 21 21 22 23 24 Achromatopsia Trolig genkälla i södra Västerbotten Samma gen kan ge mer eller mindre komplett achromatopsia 25 25 25 Hereditär pyrofosfatartropati Varierande debutålder och expressivitet Trolig genkälla i Kristianstads län 29 31 32 Orala spaltdefekter Epidemiologisk fall-referent-metodik Många barn med läpp-käk-gomspalter i Skellefteåregionen Förhöjd frekvens spalter pga gener från norr Ny metod - teoretiska tillämpningar 33 33 33 36 36 Blodgruppen "lilla p" Sällsynt överallt i världen utom i Västerbotten Genkälla i Skråmträsk 39 39 41 Tapetoretinala degenerationer Många varianter Genkälla i Burträskregionen Gemensam anfader med "lilla p" familjer 43 43 44 46 Publikationer 49 Abstracts till vetenskapliga konferenser 61 Övriga referenser 67 3 INLEDNING I min forskningsverksamhet har jag ägnat mig åt två huvudområden: A. Medicinsk genetisk forskning med utgångspunkt i genealogisk metodik. B. Forskning kring genetiska risker och miljörisker för ofödda barn. I den här skriften sammanfattas genomförda projekt inom det första av dessa områden. Det andra planerar jag att presentera i en kommande rapport från inst. for miljö- och hälsoskydd. Efter ett inledande kapitel om genetisk forskning och genetisk genealogi presenteras olika projekt i den ordning de initierats. Fortlöpande hänvisas till bifogad förteckning över publicerade rapporter (med ordningsnummer inom parentes) samt abstracts till föredrag vid vetenskapliga konferenser. Till sist följer en förteckning över ytterligare referenser som nämnts i texten. Ett arbete (9), där genealogisk metodik använts för forskning kring benmärgsinflammation (osteomyelitis) med blåsor (pustulosis) i handflator och på fotsulor (palmo-plantaris), gav inga hållpunkter för genetiska samband och kommenteras därför inte ytterligare i denna rapport. De sex projektområden som presenteras är följande: - Hereditär maculadegeneration - ärftligt betingad degeneration av gula fläcken (macula lutea) i ögats näthinna. Termen hereditär (ärftlig) används för att skilja denna typ av sjukdom från icke ärftliga varianter. - Achromatopsia - total färgblindhet med starkt nedsatt syn (amblyopia), ljuskänslighet och ryckiga ögonrörelser (nystagmus). - Hereditär pyrofosfatartropati - en ärftligt betingad reumatisk sjukdom med utfällning av pyrofosfatkristaller i leder, särskilt knäledernas menisker. Även här förekommer icke ärftliga varianter av liknande sjukdomstillstånd. - Blodgruppen "lilla p" - en mycket ovanlig blodgrupp som förekommer i oväntat hög frekvens i Västerbotten. - Orala spaltdefekter - spalter i munhålan (läpp-käk-gomspalter), vilka anses ha en multifaktoriell orsaksbakgrund, dvs flera olika gener och även miljöfaktorer är inblandade. - Tapetoretinala degenerationer - retinitis pigmentosa och liknande degenerativa sjukdomstillstånd i ögats näthinna. Av dessa projekt ägnas det förstnämnda största utrymmet, då forskningen hunnit längst inom detta område. Umeå i januari 1991 Stefan Nordström 5 GENETIK OCH GENETISK GENEALOGI Livets gåta Tomten i Viktor Rydbergs dikt "grubblar, fast ej det lär båta, över en underlig gåta." "För sin hand genom skägg och hår, skakar huvud och hätta 'nej den gåtan är alltför svår, nej, jag gissar ej detta'-" Det är livets gåta tomten grubblar över. Han har genom många generationer följt släktleden på gården. På natten smyger h a n in i barnkammaren för att se "husbondfolket det kära" och "de söta små". "Så har han sett dem, far och son, ren genom många leder slumra som barn; men varifrån kommo de väl hit neder? Släkte följde på släkte snart, blomstrade, åldrades, gick - men vart? Gåtan, som icke låter gissa sig, kom så åter!" När tomten till sist kommit till vila i höet på ladans loft, tänker han på svalan, som har sitt bo alldeles intill, "...till våren med blad och blom kommer hon nog tillbaka, följd av sin näpna maka. Då har hon alltid att kvittra om månget ett färdeminne, intet likväl om gåtan, som rör sig i tomtens sinne." En månstrimma lyser in på gubbens skägg, "strimman på skägget blänker, tomten grubblar och tänker". Långt borta i fjärran hör han bruset från "forsens fall". "Tomten lyssnar, och halvt i dröm, tycker sig höra tidens ström, undrar, varthän den skall fara, undrar, var källan må vara." J a g har låtit dessa tomtens funderingar inleda en populärvetenskaplig bok, "Livet - en gåta" (51). Skärningsfältet mellan skapelsetro och evolutionsteorier har jag behandlat i ytterligare en bok, "Charles Darwin - hädare eller hedersman?" (63) och några artiklar (35, 46, 59, 61, 62). Spekulationer och forskning kring ärftlighet I alla tider har människor grubblat över arvets mysterier. I den antika grekiska litteraturen finns hänvisningar till speciella egenskaper och släktdrag, och man spekulerade mycket kring h u r dessa gått i arv från generation till generation. Hippokrates (omkr 400 f.Kr.), kallad läkekonstens fader, hörde till dem som försökte förstå arvsmekanismerna hos människan. På stentavlor från en plats nära Ur i Kaldéen, varifrån Abraham utvandrade, har man hittat stentavlor med inristade hästhuvuden. Hästarna har rak, konkav eller konvex panna. En del saknar manhår, andra har uppstående eller nedliggande man. Allt detta är egenskaper som är väl kända från nutida hästavel, och som följer de arvslagar Mendel upptäckte 1865. Stentavlorna ger en antydan om att man redan för ca 6000 år sedan försökte förstå hur dessa egenskaper hos hästarna nedärvs. Till de tidigast dokumenterade exemplen på försök till analys av nedärvning hos människan hör vissa avsnitt i Talmud, judiska stadgesamlingar från omkr 500 e.Kr. Där finns påbud om att när två bröder avlidit på grund av förblödning efter omskärelse, skulle ytterligare söner till samma mor och söner till hennes systrar inte omskäras. Idag vet vi att blödarsjuka orsakas av ett recessivt anlag på X-kromosomen, vilket innebär att gossar drabbas om de ärver en X-kromosom med detta anlag från sin mor. Modern har inte själv blödarsjuka om hon i sin andra X-kromosom har ett dominant anlag för normal blodlevring. Hennes döttrar får inte heller blodar- 6 sjuka, då de ärver en normal X-kromosom från sin far. J u d a r n a tycks ha varit medvetna om den stora risken att bröder till redan drabbade gossar, och även söner till moderns systrar, skulle få blödarsjuka. Med empirismen från 1600-talet och framåt följde att man mer ingående började studera även livet och livsbetingelserna. Man började då också mer seriöst granska företeelser som nedärvning och variation i mänskliga populationer. Redan 1772 beskrev Maupertuis en familj där polydaktyli (extra fingrar eller tår) uppträdde i fyra på varandra följande generationer. All forskning på 1600-talet var kanske ändå inte så seriös. När Leuwenhoek konstruerat de första användbara mikroskopen tittade man på diverse celler. I sädesvätska från tjurar sade man sig då ha sett mikroskopiska men helt färdiga kalvar. I sädesvätska från tuppar tyckte man sig se alldeles färdiga små kycklingar som flaxade med sina vingar. Det publicerades också bilder av spermier från människa, i vilka man kunde se ett litet miniatyrbarn inne i spermiehuvudet. Dessa resultat från mikroskoperingens barndom vittnar inte bara om att man hade ganska dåliga mikroskop, u t a n även om manlig fåfänga. Det var män som studerade livet, och de ville nog gärna framhäva sin egen betydelse i fortplantningen. Det var faktiskt inte förrän 1875 som man blev riktigt på det klara med att befruktningens egentliga väsen är sammansmältning mellan en hanlig och en honlig cell, mellan en spermie och ett ägg. Då hade det gått tio år sedan Mendel upptäckt att det finns bestämda arvsenheter, som överförs från generation till generation. Mendel införde begreppen dominant respektive recessivt arv. Redan 1814 publicerade dock Joseph Adams en skrift (A Treatise on the Supposed Heredity Properties of Disease), där h a n tycks ha gjort åtskillnad mellan dominant och recessivt arv genom att använda termerna "hereditary" respektive "familial". På Mendels tid var flertalet forskare ä n n u inte mogna att förstå och acceptera hans ärftlighetslagar. Man tyckte att hans statistiska beräkningar mer liknade talmystik ä n seriösa försök att förstå arvets ^mysterier. År 1900 återupptäcktes emellertid Mendels lagar av tre forskare, vilka arbetade oberoende av varandra (de Vries i Holland, Correns i Tyskland och von Tschermak i Österrike). Kort tid därefter hade man klart för sig att dessa lagar gäller även för människan. Den engelske läkaren Garrod visade 1902 att ämnesomsättningssjukdomen alkaptonuri följer en recessiv nedärvning enligt Mendels modell. Några år senare (1909) påvisade h a n i skriften "Inborn errors in man" mendelsk nedärvning för flera olika ämnesomsättningssjukdomar. Vad Mendel upptäckte var i korthet följande: - Det finns någon form av enskilda arvsanlag, som kan gå i arv från generation till generation. Tidigare hade man allmänt trott på något slags "blandningsgenetik", enligt vilken kroppsvätskor och vävnadsfragment blandades (jämför uttiyck som "halvblod" och "helblod"). - Anlagen finns i parvis uppsättning och skiljer sig åt vid bildning av könsceller (gameter). - Anlagen fördelas slumpmässigt på gameterna. Sedan 1960 har Mc Kusick (senaste upplaga 1990) gett u t en katalog över autosomalt dominanta, autosomalt recessiva och Xkromosombundna fenotyper hos människan. Med fenotyp menas det sätt på vilket anlagen (genotypen) kommer till uttiyck. 7 Autosomer kallas de 44 (22 par) kromosomer som inte är könskrornosomer. Autosomal innebär att anlaget sitter på någon av dessa. Det 23:e paret bestämmer kön. Eftersom könskromosomerna är lika hos flickor (XX) och olika hos pojkar (XY) är det viktigt att skilja mellan könskromosombundet arv och autosomalt arv. Vi känner för närvarande inte till några Y-bundna sjukdomar, däremot ett flertal X-bundna. Genetik - Genesis Genom att Mendels ärftlighetslagar återupptäcktes år 1900, kan man säga att det blev genetikens egentliga födelseår. Termen genetik (genetics) lanserades emellertid först 1905 av den engelske genetikern Bateson. En dansk ärftlighetsforskare (Johannsen) lär vara den som först använde termen gen som benämning på arvsanlag (1911). Termen genetik, som bygger på samma grundord som Genesis, benämning på Första Moseboken i Bibeln, är ett passande namn på denna vetenskapsgren. Skapelseberättelsen och ärftlighetsforskningen har båda, var på sitt sätt, med livets ursprung och innersta hemligheter att göra. De har alltså samband med tomtens funderingar kring "en underlig gåta". På 1800-talet böljade man förstå att arvet är kopplat till kromosomerna i cellkärnan. Redan i slutet av det århundradet försökte man i vävnadssnitt avgöra hur många kromosomer som finns i människans celler. Inte förrän 1956 lyckades man emellertid med säkerhet fastställa människans kromosomantal till 46. Det var två genetiker verksamma i Lund, Tjio och Levan, som lyckades med den bedriften genom att göra preparat på celler odlade i vävnadskultur. I dag är det rutin på klinisk genetiska laboratorier att göra sådana kromosompreparat för att bl.a. påvisa kromosomavvikelser, av vilka den mest kända, en tredubbel uppsättning av kromosom nr 21 (trisomi 21), orsakar Down's syndrom ("mongolism"). År 1902 framförde W.S. Sutton teorin att anlagen (generna) är belägna i kromosomerna. Under senare delen av 1900-talet har det skett en explosionsartad kunskapsutveckling på genetikens område. Watson och Crick presenterade 1953 en modell för strukturen hos DNA (deoxyribonucleic-acid), arvets molekyl (på svenska: deoxyribonukleinsyra). Från böljan av 1960-talet har vi förstått h u r denna märkliga molekyl kan fungera som genetisk kod. DNA i cellkärnan kopieras till budbärar-RNA (ribonukleinsyra), som sedan i cellplasmans ribosomer utgör mall för cellens proteinsyntes. Under de två senaste decennierna har vi lärt oss behärska genteknik, som inkluderar möjligheter att kartlägga enskilda gener, ställa sjukdomsdiagnos på molekylär nivå och behandla vissa sjukdomar med genterapi, dvs ersätta felaktiga gener med normalt fungerande. Det första egentliga försöket med genterapi på somatiska celler (kroppsceller) hos människan genomfördes i USA i slutet av förra året (1990). Genen för ADA (adenosindeaminas) överfördes då till spädbarn, som genom en ärftlig defekt saknar förmåga att bilda detta för immunförsvaret viktiga enzym. Sjukdomen kallas SCID (severe combined immuno deficiency). Modern genteknik ger oss fortlöpande allt större kunskap om grundläggande orsaksmekanismer bakom ärftliga sjukdomar. Därigenom får vi också bättre och bättre möjligheter att i framtiden k u n n a bota sådana sjukdomar. 8 Den snabba kunskapsutvecklingen på det biomedicinska området innebär att etiska problem accentuerats. En del av dessa problem har jag diskuterat i boken "Manipulerad produkt eller välsignad livsfrukt" (57) och i några andra skrifter och artiklar (45, 53, 60). I nämnda bok och även "Livet - en gåta" (53) behandlas viss grundläggande genetik. Mer omfattande läroböcker om människan och arvet har publicerats av Beckman, 1983 samt Lindsten och Iselius (red.), 1987. Genetisk genealogi I begreppet genealogi finns en liknande grekisk ordstam (genea = härkomst) som i genetik och genesis (gennao = ursprung, alstra). Genealogi (släktforskning) är en viktig hjälpvetenskap inom medicinsk genetik. Hos historiskt kända personer har man ibland k u n n a t följa ärftliga sjukdomar eller egenskaper under mycket lång tid. Lättast är det av naturliga skäl att följa dominanta arvsanlag, eftersom de kan komma till uttiyck i generation efter generation. Vem minns inte historieböckernas porträtt av medlemmar av den habsburgska furstedynastin med den karakteristiskt utskjutande underläppen? Det draget fanns med hos kejsar Maximilian I (född 1459) och återfinns sedan hos dennes ättlingar i rakt nedstigande led, t ex kejsar Karl V, Maria Theresia av Österrike och kung Alfonso av Spanien. Ett annat exempel på en egenskap som kunnat följas genom många generationer är synfalangi (sammanvuxna fingerleder), som nedärvs autosomalt dominant. I samband med en gravöppning 1874 kunde man påvisa denna defekt hos förste earlen av Shrewsbuiy, född 1390 (omnämnd av Shakespeare i "Henrik VI"). En likadan skelettdefekt har återfunnits hos en ättling 14 generationer senare (A.H.Talbot) samt dennes far och farfar. En välkänd "historisk" gen är också det tidigare nämnda X-kromosombundna anlaget för blödarsjuka, som kunnat härledas till bl.a. drottning Viktoria av England. Det är inte så vanligt att man kunnat följa ärftliga sjukdomar och egenskaper så många generationer bakåt i tiden. I Sverige har vi emellertid större möjligheter än i andra delar av världen att bedriva omfattande genetisk genealogisk forskning genom vår tillgång till väl bevarat historiskt källmaterial, främst husförhörslängder och andra kyrkoarkivalier. Flera avhandlingar, där genealogisk metodik ingått som ett viktigt led i arbetet, har publicerats vid svenska universitet. Som exempel kan nämnas en doktorsavhandling av Rune Andersson (1976) om familjär amyloidos med polyneuropati (den s.k. "Skellefteåsjukan"). Andersson beskrev denna sjukdom hos 60 patienter från norra Sverige. En utbredningskarta visar att 37 av dessa var födda i norra Västerbotten med koncentration till området kring Skellefteå. Omfattande forskning kring denna sjukdom bedrivs n u av Ulf Drugge vid Forskningsarkivet, Umeå universitet, i samarbete med bl.a. Gösta Holmgren (klinisk genetik). Sjukdomen följer en autosomal dominant nedärvning, men det är inte säkert att alla som bär anlaget verkligen blir sjuka (ofullständig penetrans). Namnet amyloidos kommer av att det i vissa vävnader, bl a perifera nerver, avlagras stärkelseliknande degenerationsprodukter (amylon = stärkelse), som kan ge störningar i nervfunktioner. Debutåldern för symtom bland de 60 patienterna i Anderssons avhandling varierade mellan 29 och 75 år. Sjukdomen förekommer nästan dubbelt så ofta hos män som hos kvinnor. 9 Även autosomalt recessiva varianter av sjukdomen familjär amyloidos förekommer. Av den autosomalt dominanta varianten ("Skellefteåsjukan") är n u ett hundratal fall kända i Sverige, men sjukdomen är känd även i t. ex. Portugal. Man vet idag att den orsakas av en mutation i genen för prealbumin, som sitter i kromosom nr 18, och sjukdomen kan diagnosticeras med genteknik. En a n n a n välkänd "Västerbottenssjukdom" är Sjögren-Larssons syndrom, som beskrivits i en avhandling av Sten Jagell (1981). Av 58 identifierade svenska patienter, födda 1886-1978, var 45 födda inom ett begränsat område kring Skellefteå-Piteå. Ytterligare fem hade anfäder inom detta område. Sjukdomen karakteriseras av medfödda hudförändringar, förlamningar i armar och ben samt psykiskt utvecklingshandikapp, och följer en autosomal recessiv nedärvning. Jagell har konstruerat pedigrees över denna sjukdom, som går tillbaka till senare delen av 1600-talet. Vad ett pedigree är förklaras på sid. 11. Akut intermittent porfyri är en ämnesomsättningssjukdom som beskrevs av lappmarksdoktorn Einar Wallquist redan på 1930-talet, och som granskats i en doktorsavhandling av Lennart Wetterberg (1967). Beckman och Bergenholtz (1975) har påvisat en trolig genkälla för den svåra hudsjukdomen epidermolysis bullosa i Burträskregionen. Holmgren et al. (1988) och Drugge et al. (1990) har med genealogisk metodik påvisat ärftlighet för mental retardation betingad av en strukturell defekt på X-kromosomen. Andra intressanta genetiska studier där kyrkoarkivalier kommit till användning är en kartläggning av utbredning och arvsgång för Morbus Gaucher, en ärftlig rubbning av fettomsättningen, (Hillborg och Sundström, 1979), samt en omfattande kartläggning av schizofreni i ett nordsvenskt isolat (Böök et al., 1980). Drugge (1988) har i en Scriptum-rapport skrivit om husförhörslängder som medicinsk urkund vid studier av psykiska sjukdomar och förståndshandikapp. Populärvetenskapliga artiklar om genetisk genealogisk forskning har publicerats i Forskning och Framsteg (8, 33) och i ett par andra skrifter (49, 54). 11 HEREDITÄR MACULADEGENERATION Centrala delen av ögats näthinna (macula lutea, gula fläcken) drabbas lätt av degenerationer, vilka medför att detaljseendet avtar eller försvinner. Flera hereditära (ärftliga) varianter av denna sjukdom är kända, såväl dominanta som recessiva. Maculadegenerationer kan även uppträda som åldersförändringar eller som effekt av mycket starkt ljus mot ögat (t ex hos svetsare som inte skyddat sina ögon tillräckligt), exponering för droger, eller a n n a n exponering. Redan 1905 beskrev en tysk läkare (Best) en autosomal dominant form av hereditär maculadegeneration (HMD) hos 8 av 59 undersökta medlemmar av en familj. Sedan dess har flera pedigrees med samma typ av sjukdom, som därför ibland kallas Bests sjukdom, presenterats i såväl europeisk som amerikansk litteratur. Ett pedigree är ett slags släkttavla som visar nedärvningsmönster. Som symboler brukar m a n använda cirklar för flickor/ kvinnor och fyrkanter för gossar/män. Proband (ibland propositus) kallas den person från vilken släktutredningen utgår. Generationer kan betecknas med romerska och individer med arabiska siffror. I det följande presenteras flera exempel på sådana pedigrees. En svensk ögonläkare, Yngve Barkman, presenterade 1961 en stor familj från Dalarna med 69 diagnosticerade fall av en ögonsjukdom, som han kallade central tapetoretinal degeneration. Vi kunde senare visa att det rörde sig om HMD av typen Bests sjukdom (se nedan). Mitt eget "äventyr" med HMD böljade när jag i slutet av 60-talet sökte efter ett lämpligt ämne för en 3-betygsuppsats i ämnet genetik. J a g fick då kontakt med barnläkaren Gösta Holmgren, n u överläkare i klinisk genetik, och ögonläkaren William Thorburn vid regionsjukhuset i Umeå. De kände till två familjer med HMD i Västerbottens län. Min uppgift blev att undersöka om dessa familjer hörde samman genealogiskt och vilket slag av ärftlig maculadegeneration de drabbats av. Mitt 3-betygsarbete presenterades i samband med en internationell konferens i Uleåborg, Finland, 1971 och ett sammandrag publicerades året därpå i Acta Ophthalmologica (1). I en av de båda västerbottniska familjerna kunde vi diagnosticera sjukdomen hos 24 av 43 oftalmologiskt undersökta familjemedlemmar. Samtliga kunde genealogiskt knytas till en anfader född 1704 (fig.l). I den andra familjen fann vi 6 fall av HMD (fig.2). I båda familjerna kunde sjukdomen konstateras i minst tre på varandra följande generationer och med relativt j ä m n fördelning mellan könen, vilket talade för en autosomal dominant nedärvning. HMD och husförhör I mitt 3-betygsarbete kunde jag inte visa att de båda familjerna hörde samman. 1:1 i fig.2 var hittebarn från Stockholm, och hade som ettåring hämtats upp som "drängämne" till en by i södra Lappland. Han blev kyrkobokförd som far till 11:2, men släktingar i senare generationer hävdade bestämt att en resande, som tillfälligt besökt byn (1:2), var den biologiske fadern. Man hävdade också bestämt att det var denne resande som fört genen för HMD in i familjen. 12 9 @ ® O O d • W • • p Diagnosticerad HMD Proband +/ M i s s t ä n k t HMD O f t a l m o l o g i s k t undersökt Troligen HMD ' Dog relativt ung Inga maculadegenerationer eller synproblem kända N o t e r i n g i h u s f ö r h ö r s l ä n g d som indikerar synprobiem Fig.l. Pedigree över familj 1 med 24 fall av HMD. 13 rf ? B 0 W % • O • o + Diagnosticerad HMD Misstänkt HMD Trolig HMD Inga m a c u l a d e g e n e r a t i o n e r eller synproblem kända Oftalmologiskt undersökt Fig.2. Pedigree över familj 2 med 6 fall av HMD. När jag arbetade med släktutredningar i de båda familjerna kom jag i kontakt med Egil Johansson (Scriptums redaktör), som berättade om sin forskning kring husförhörslängder. Det slog mig då att en närmare granskning av läsbetyg, betyg i begrepp och förstånd samt noteringar i anmärkningskolumner i dessa längder, skulle kunna ge vägledning i mina försök att spåra genen för HMD bakåt i tiden. Det visade sig också att flera av anfäderna i fig. 1 hade fått svaga läsbetyg, även om de hade bra betyg i begrepp och förstånd. Någon gång hade prästen, kanske som förklaring till det dåliga läsbetyget, skrivit "svagsynt" i anmärkningskolumnen. Ett P vid symbolen för vissa anfäder markerar att det finns noteringar i husförhörslängderna som indikerar svagsynthet. P står for ett försök att översätta "husförhörslängder" till engelska (PCMER = "parish catechetical meeting examination records"). Genen för HMD tycks sålunda ha kommit in i denna släktgren genom 111:5 (fig.l), som gifte sig med en sonson till anfadern född 1704. Inget av dennes barn eller barnbarn hade, av husförhörslängderna att döma, några problem med synen. Fig.3 är hämtad ur en artikel i Forskning och Framsteg, som jag publicerat tillsammans med Egil Johansson (8). Figuren visar nämnda samband med inlagda symboler för läsbetyg. En intressant notering var att såväl IV: 1 som IV:7, som båda tycks ha fört sjukdomen vidare, fick sina läsbetyg ändrade från "läser svagt" till "läser någorlunda" i samband med giftermålet. 14 Fig.3. Illustration som visar symboler använda i husförhörslängder samt noteringar i dessa längder för anfäder de sex första generationerna i ett utvidgat pedigree över fam. 1 (fig. 1). 15 Genkälla i Vilhelmina, Västerbottens län Genom ytterligare undersökningar i den första familjen kunde vi utöka antalet diagnosticerade fall av HMD till 35 (2), som alla kunde knytas till "kvinnan med det dåliga läsbetyget" (111:5 i fig. 1 och 3). Då det av husförhörslängderna framgick att hon kunde vara den som fört in genen för HMD i denna släkt vidtog ett omfattande "detektivarbete" för att spåra hennes anor. Det visade sig vara mycket svårt. Hon hade varit mycket rörlig, dvs flyttat mellan olika byar och församlingar. Hennes födelseår liksom hennes n a m n (ibland Christina ibland Kjersten) varierade från källa till källa. Så småningom gick det dock att slå fast att hon var nybbyggaredotter från Hacksjö i Vilhelmina. Hon var tredje barnet bland åtta syskon (fig. 4) och med ytterligare genealogiska utredningar kunde jag visa att en äldre bror och en yngre syster också hade fört genen för HMD vidare. Tre pedigrees över denna 'Vilhelmina-familj", ett från vardera av de tre nybyggarebarn som fört genen vidare, kunde nu publiceras (3). Fig.4. Nybyggareparet EO och MA i Hacksjö, Vilhelmina, med åtta barn, av vilka tre fört genen för HMD vidare till senare släktled. Här presenteras ett av dessa tre pedigrees (fig. 5), nämligen det som utgår från det näst äldsta nybyggarebarnet (född 1781). I detta pedigree ingår den andra av de familjer som jag ursprungligen studerade (fig.2). Misstanken att sjukdomen förts in i denna familjegren genom illegitimitet kunde sålunda avfärdas. I stället var det modern i generation I som fört genen för HMD vidare och hon var alltså ättling till nybyggarna i Vilhelmina. I fig. 5 och följande pedigrees har även symbol för konvergent (inåtriktad) skelning lagts in. Sådan skelning förekommer ofta bland medlemmar av Älvdalen- och Vilhelmina-familjen, men det kan ha delvis andra ärftliga orsaker och behöver sålunda inte vara kopplat till HMD. 16 /? • O Diagnosticerad HMD / M i s s t ä n k t HMD / ' Sekundär p r o b a n d Trolig HMD Kl fe K o n v e r g e n t skelning • O + Primär p r o b a n d Oftalmol._undersökt t Dog r e l a t i v t u n g Maculadegenerationer eller synproblem ej kända Fig.5. Pedigree över släktgren från n ä s t äldsta barnet (f.1781) till nybyggareparet i Hacksjö, Vilhelmina. 17 De båda ursprungliga familjerna med HMD kunde alltså slutligen sammanföras i en enda, och antalet diagnosticerade fall utökades till 125 (65 män och 60 kvinnor), av vilka 23 var döda och 27 hade flyttat ut från Västerbottens län. För att nå detta resultat krävdes en mycket omfattande genomgång av ögonjournaler och olika register över synskadade i länet, kombinerad med mycket intensiva genealogiska efterforskningar och förnyad synundersökning av ett stort antal "misstänkta fall". Ytterligare 22 fall av HMD i länet upptäcktes genom denna granskning. Dessa visade sig dock representera andra varianter av HMD, såväl dominanta som recessiva. Diagnos och arvsgång för dessa diskuteras utifrån sammanlagt nio mindre pedigrees (3). År 1974 sammanfattade jag mina då tillgängliga forskningsresultat i en doktorsavhandling om HMD i Västerbottens län (5). I denna avhandling ingår även en biokemisk studie (4). Vissa biokemiska avvikelser hos patienter med autosomal dominant HMD hade tidigare rapporterats av en amerikansk forskare. Någon sådan avvikelse kunde inte påvisas i vårt familjematerial. I en senare amerikansk forskningsrapport bekräftades att de aktuella amerikanska patienterna hade normala EOG-värden (se nedan), vilket visar att de har en a n n a n variant av HMD ä n den som förekommer i det stora västerbottniska familjematerialet. Primär genkälla i Älvdalen, Kopparbergs län Nybyggaren i Hacksjö var dalmas. J a g återfann honom i "Gamla byar i Vilhelmina" (Pettersson, 1941), med uppgiften att h a n gift sig med en samekvinna. Sjukdomen är dock inte känd bland samer, och med tanke på att Barkman (1961) beskrivit en liknande sjukdom i Dalarna (se ovan), låg det nära till hands att misstänka att dalmasen, var den som kommit in med genen för HMD i den västerbottniska populationen. En granskning av källorna (husförhörslängder, flyttlängder och dopböcker) visade dock att Pettersons bok innehåller felaktiga uppgifter. Nybyggaren hade med sig en dalkulla, när han med deras förstfödde slog sig ner i Hacksjö. Hon blev mor till alla hans åtta barn. Genom att kontakta Dr Barkman kunde jag få tillgång till hans källmaterial, och med det begav jag mig till Dalarna, där jag inventerade förekomsten av alla nyupptäckta fall med den form av "central tapetoretinal degeneration", som Barkman tidigare beskrivit. Vi kunde bekräfta att det rörde sig om HMD av samma slag som vi beskrivit i Västerbotten, och så småningom kunde vi knyta 116 fall av HMD (63 män och 53 kvinnor) till ett stort pedigree från Dalarna (6). Samtliga var ättlingar till ett par födda i byn Kittan i Älvdalens församling 1677 resp. 1681 (fig.6). Efter mycket intensivt sökande återfann jag, i husförhörslängd för Älvdalens församling 1775-85, nybyggareparet från Vilhelmina och deras förstfödde, då bosatta i byn Klitten. Rätt födelseår för de båda makarna var 1747 respektive 1760. (I kyrkböcker från Vilhelmina sägs mannen vara född omkr. 1745 och kvinnan 1759). I kolumnen för "bortflyttade" finns noterat "Ångermanland". Omkring 1780 hade de dragit sina färde, sannolikt först ner mot kusten, för att sedan följa Ångermanälven upp mot Hacksjö i Vilhelmina. 18 Fig.6. Pedigree över Älvdalen-släkten med 116 fall av HMD. I övre högra hörnet ser vi hur Vilhelmina-släkten genealogiskt kan kopplas till Älvdalen-släkten. (Teckenförklaring, se nästa sida.) Diagnosticerad HMD T r o l i g HMD K o n v e r g e n t skelning Maculadegenerationer eller synproblem ej kända Primär proband Sekundär proband O f t a l m o l o g i s k t undersökt Dog relativt ung 20 Nybyggarehustrun var sonsondotter till anfäderna från byn Kittan, och det var hon som förde genen för HMD in i den västerbottniska populationen. Som n ä m n t s ovan menade m a n i en av släktgrenarna i Västerbotten (fig. 2) att HMD hade kommit in i släkten via en tillfällig resande, som uppehållit sig i en by i södra Lappland. I Dalarna var det faktiskt flera familjemedlemmar som hävdade att HMD kommit in i deras släkt via en iyss, som någon gång på 1800-talet uppehållit sig där! Som vi sett fanns emellertid genen för HMD i Dalarna redan på 1600-talet. Vi vet inte om det var m a n n e n eller h u s t r u n i byn Kittan, som var anlagsbärare. HMD-genen k a n ha uppstått hos någon av dem genom mutation. Hade denna gen funnits i populationen i tidigare generationer, bör den ha k u n n a t sprida sig till fler släktgrenar. Nu tycks det vara så att så gott som alla i Sverige, som har j u s t denna variant av HMD, på ett eller annat sätt kan knytas till de n ä m n d a anfäderna. Genen har förts vidare till senare generationer via tre av deras sex barn, av vilka de båda förstfödda var tvåäggs tvillingar (fig. 7). Fig.7. Genealogisk länkning mellan Älvdalen- och Vilhelminasläkten Genen har "utvandrat" till USA I dag h a r betydligt mer än 250 kända fall av HMD i Sverige (mer än 120 i Älvdalen-gr enen och mer ä n 130 i Vilhelmina-grenen) kunnat knytas till genkällan i Kopparbergs län. Många av de patienter som ingick i tidigare undersökningar har avlidit, men ständigt upptäcks nya fall i senare generationer. En uppföljande studie av sjukdomens spridning i Älvdalen- och Vilhelmina-släkten pågår. I Älvdalens församling bor idag sammanlagt 27 personer med HMD, födda 1915 eller senare, vilka samtliga h a r sina rötter hos anfäderna i byn Kittan på 1600-talet. 21 Genen för HMD har också "utvandrat" till Minnesota i USA, där åtminstone ett 20-tal fall är kända. J a g fick detta bekräftat vid ett besök vid University of Minnesota samt Mayo-kliniken i Rochester sommaren 1985. Genom medlemmar i Älvdalen-familjen har jag fått uppgifter om släktingar med HMD, som utvandrat till Minnesota. Varierande symtombild Genen för HMD ger i detta stora svenska familjematerial mycket varierande symtombild (varierande expressivitet). Det enda som är gemensamt är att gula fläcken förr eller senare drabbas av degeneration, vilket leder till försämrat detaljseende. I en publikation i Läkartidningen (7) finns färgbilder som visar exempel på h u r maculaområdet kan se u t hos olika patienter. Yngre debutanter får ofta en gulaktig, blåsliknande cysta i maculaområdet. Den kan likna en äggula, varför sjukdomen ibland kallas vitelliform (vitellus är latin för äggula). Senare kan denna "äggula" torka ut och "brytas sönder" med kraftiga pigmentförskjutningar och pigmenthopklumpningar som följd. Detta stadium kallas därför ibland "vitelliruptive" i engelskspråkig litteratur. Det är dock sannolikt så, att vitelliforma stadier aldrig uppträder hos en del patienter, där man i stället ser mer eller mindre diskreta pigmentförskjutningar och uppklarningar centralt i näthinnan. Somliga drabbas av mycket starkt nedsatt detaljseende, även om degenerationen oftalmoskopiskt kan förefalla ganska måttlig, medan andra kan ha kvar relativt bra synskärpa trots markanta pigmentförskjutningar. Genbärare spåras med EOG Gemensamt för alla som har denna form av HMD är att de har patologiska EOG-värden. Med EOG avses elektrooculografi. Ögat har en positiv pol vid hornhinnan och en negativ vid näthinnan. Normalt ökar spänningsskillnaden mellan dessa poler när ljus faller mot ögat. Med känsliga instrument kan man avläsa detta via små plattor som fästs vid näsroten respektive ytterkanten av ögat. Genom att låta patienten vrida blicken fram och tillbaka mellan två fixeringspunkter kan man mäta hur den positiva polen vid hornhinnan förskjuts mellan dessa punkter. J u större spänningspotentialen är, j u större blir utslaget. Vid EOG-undersökningar mäter man kvoten mellan ljusvärden och mörkervärden. Dessa värden varierar hos olika personer, men normalt ligger ljus/mörker kvoten någonstans mellan 1,6 och 4. Hos patienter med den form av HMD som finns i Vilhelmina- och Älvdalen-släkten ligger den dock alltid mycket nära 1,0, vilket visar att näthinnan hos dessa inte aktiveras på normalt sätt när ljus faller mot ögat. I samarbete med William Thorburn och ögonkliniken i Umeå har vi visat att inte bara kliniskt drabbade, u t a n även sådana som är genbärare men ä n n u inte debuterat med nedsatt syn eller maculadegeneration, har patologiska EOG-värden (10). Bland kliniskt friska barn till förälder med diagnosticerad HMD, hade flertalet normalt EOG, men några visade patologiska värden. När dessa senare, som kan betraktas som anlagsbärare före debut, räknades samman med de barn som redan debuterat, utgjorde de tillsammans exakt 50 procent, vilket man skall förvänta sig vid autosomalt dominant arv. 22 Flera av dessa till synes kliniskt friska barn med patologiskt EOG har n u debuterat med mer eller mindre diskreta pigmentförskjutningar eller vitelliforma stadier av maculadegeneration. • • Hereditär maculadegeneration (HMD) H @ Anamnestisk synnedsättning - sannolikt HMD • © Sannolikt HMD " S ^ Konvergent skelning OftaImologiskt undersökt Fig.8. Gren av Vilhelmina-släkten med HMD. Homozygota anlagsbärare I Vilhelmina-familjen hade två bröder sex respektive sju barn, som samtliga hade diagnosticerad HMD, Bröderna var sannolikt homozygota för HMD-genen, dvs de hade ärvt denna gen från både sin far och sin mor (fig.8). Genen överförs då till samtliga barn. I Älvdalen-familjen har en m a n elva barn (fig. 9). Av dessa har tio, samt två barnbarn, diagnosticerad HMD. Synnedsättning debuterade i barnaåren (3-9 år). Hos en av döttrarna kunde Barkman konstatera maculaförändringar före ett års ålder. 23 Fig.9. Gren av Älvdalen-släkten med HMD. Teckenförklaring, se fig.8. Med EOG-undersökning var det möjligt att konstatera att även den dotter, som ä n n u vid 27 års ålder inte debuterat med maculaförändringar eller synnedsättning, är bärare av genen för HMD, vilket senare bekräftats av att en femårig son till henne fått ganska markanta degenerationer i maeulaornrådet. Fadern till de elva barnen är alltså med mycket stor sannolikhet homozygot för den dominanta genen (22), dvs h a n har den i dubbel uppsättning och har ärvt den från både sin far och sin mor. Det är mycket ovanligt att m a n hittar patienter med homozygot uppsättning av en dominant sjukdomsgen. Även farfadern och en äldre syster till honom kan ha varit homozygota (fig.9). En uppföljande studie "pågår i den homozygote faderns familjegren, och antalet barnbarn har n u ökat till elva. För vart och ett av dessa barnbarn är risken 50 procent att de skall drabbas av HMD. Varierande debutålder Som framgått ovan kan debutåldern variera högst avsevärt. I litteraturen har det sagts att sjukdomar av detta slag gärna debuterar i samband med mer eller mindre "dramatiska" händelser i kroppen, t ex puberteten. I diagrammet (fig. 10), där åldersgränserna delats upp i snäva intervall j u s t kring pubertetsåldrarna, finner vi dock att det är mycket få som debuterat i dessa åldrar. Även om vi delar u p p debutåldrarna i j ä m n a femårsintervall visar de en tvåpucklig fördelning, som tyder på debut antingen före eller efter puberteten. Av någon anledning, som vi inte kan förklara, är det vanligare att flickor debuterat före och pojkar efter puberteten. Dessa skillnader är statistiskt säkerställda. Som synes förekommer debutåldrar ända upp i 40-50-årsåldern. 24 Fig.lO. Stapeldiagram som visar hur debutåldern för synnedsättning varierar i Älvdalen- och Vilhelminasläkten. Försök att isolera och molekylärt definiera genen for HMD Medicinska forskningsrådet har beviljat anslag på sammanlagt 1,25 milj. kr för perioden 1/1 -91 - 3 0 / 6 -93 till ett projekt vid Umeå universitet med målsättningen att fastställa den kromosomala lokalisationen för gener som orsakar HMD och en del andra ärftliga sjukdomar, som är vanliga i norra Sverige. Försök skall göras att isolera genen för att därigenom närmare kunna studera dess funktion. Möjligheter att isolera gener för ärftliga sjukdomar är av stor betydelse för säkrare diagnos och genetisk information. Om man närmare kan karakterisera genens avvikande funktion kan det leda till möjligheter att bota sådana sjukdomar i framtiden. Vår forskning kring HMD i Sverige har rönt stort intresse utomlands. Resultaten har presenterats vid internationella konferenser i Umeå (19), Reykjavik (23), Mariehamn (24) och Oslo (abstract publicerat i Clinical Genetics 13:131-132, 1978). Under en resa i USA 1985 föreläste jag om denna forskning vid University of Pittsburgh och University of Minnesota, samt vid Mayo-kliniken i Rochester. Resultat från vår forskning kring HMD har fortlöpande redovisats i samband med medicinska riksstämman i Stockholm (1971, 1974, 1976, 1977 och 1980, se förteckning över publicerade abstracts). 25 ACHROMATOPSIA I ögats näthinna finns två olika typer av synsinnesceller, stavar och tappar. Av de sistnämnda finns det tre varianter, vilka registrerar färger i olika våglängdsområden. De mest kända varianterna av ärftlig färgblindhet drabbar endast vissa typer av tappar, t ex rödgrönfärgblindhet. Anlag för denna form av färgblindhet sitter i Xkromosomen. En mycket ovanlig och därför mindre känd färgsinnesdefekt är total färgblindhet (achromatopsia) med starkt nedsatt synförmåga (amblyopia). Patienter som drabbas av typisk achromatopsia får en synskärpa omkring 0,1 bilateralt, små darrande eller ryckiga ögonrörelser (nystagmus), särskilt i barndomen, och blir mycket ljuskänsliga. Färgblindheten kan dock vara mer eller mindre komplett, och man har diskuterat om det kan röra sig om olika gener, som ger upphov till olika varianter av sjukdomen. I samarbete med Werner Polland, då verksam vid ögonkliniken i Umeå, har jag studerat två västerbottens-familjer med achromatopsia. I den ena (Fam. 1, fig. 11) hade fyra av sex syskon drabbats, i den andra (Fam.2, fig. 11) tre av fyra syskon. I samband med undersökning av den drabbade syskonskaran i Fam. 1 fick vi kännedom om att en kusin (111:14), bosatt i en a n n a n del av Sverige, hade samma typ av symtom som de fyra syskonen. Ytterligare ett fall, en äldre släkting boende i samma region som de drabbade syskonen, upptäcktes i samband med de genealogiska efterforskningarna (VIII:5,fig. 12). Trolig genkälla i södra Västerbotten Det var inte möjligt att föra samman de två familjerna med achromatopsia till någon gemensam anfader. Resultaten visar dock att det sannolikt fanns en genkälla för denna sjukdom någonstans i södra Västerbotten eller gränsområdet mellan Västerbottens och Västernorrlands län, där gemensamma anfäder till patienternas föräldrar var bosatta i närliggande byar på 1600-talet (fig. 13). Nedärvningen är autosomalt recessiv. Med hjälp av mycket känsliga färgsinnestest kunde vi påvisa en svag manifestation av genen även hos vissa anlagsbärare med en enkel (heterozygot) uppsättning av genen (streckade symboler ifig. 11 och 12). En son till en heterozygot anlagsbärare, kusin till de drabbade syskonen i Fam.2 (111:28, fig. 11), fick diagnosen grönblindhet (deuteranomali, deuteranopi). Samma gen kan ge mer eller mindre komplett achromatopsia Det kanske mest intressanta resultatet i detta projekt är att patienterna i de båda undersökta familjerna uppvisar achromatopsia av olika grad mellan gränserna för vad som brukar betraktas som komplett och inkomplett variant. Vi har kunnat mäta detta med hjälp av ett instrument för färgsinnesundersökningar (anomaloskop). En och samma recessiva gen tycks sålunda i dubbel dos (homozygot form) k u n n a manifestera sig i mer eller mindre komplett achromatopsia (fig. 14). Resultaten har publicerats i Acta Ophthalmologica (20) och Human Heredity (21). De har även presenterats i samband med den tidigare nämnda internationella konferensen i Reykjavik (23) och i samband med medicinska riksstämman 1979 (se abstracts). 26 Fig. 11. Pedigrees över två västerbottniska familjer med fem respektive tre fall av achromatopsia. 27 i Fig. 12. Pedigree som visar genealogiska länkningar i den första av de två familjerna i lig. 11. 28 Fig. 14. Diagram som visar att testresultat med anomaloskop för de fem fallen i F a m . l (fig. 11) varierar mellan gränserna för vad som brukar klassas som komplett och inkomplett achromatopsia. Motsvarande resultat erhölls vid testning av syskonen i Fam.2. 29 HEREDITÄR PYROFOSFATARTROPATI Pyrofosfatartropati är en reumatologisk sjukdom med förkalkningar och artritsymptom (ledinflammationer). Sjukdomen har fått sitt namn av att förkalkningarna består av pyrofosfatkristaller (kalciumpyrofosfatdihydrat). Sporadiska fall av denna sjukdom har rapporterats från hela världen. Familjära former däremot endast i europeiska länder - Tyskland, Holland, Frankrike, Tjeckoslovakien (bland ungerska ättlingar i Slovakien) och Spanien. Utanför Europa har ärftliga varianter beskrivits endast bland spanska conquistadorättlingar på Chiloeöarna utanför Chile och i familjer med europeisk härstamning i USA och Canada. I Sverige är tre familjer med denna sjukdom kända. Flertalet patienter, totalt 35 när denna studie gjordes, är bosatta i Skåne (fig. 15). Familjemedlemmar med atypiska symtom, samt yngre symtomfria släktingar, finns dock utspridda över hela södra och mellersta Sverige. Två av familjerna (A och B, fig. 16) har studerats kliniskt av reumatologerna Anders Bjelle och Asa Hagstam. Den tredje (C) har tidigare beskrivits av Ekelund. Sjukdomsbilden varierar, vilket framgår av fig. 16, där röntgenologiskt observerade förkalkningar samt kliniska symtom (artriter), typiska och/eller atypiska, markerats med olika symboler. fosfatartropati (Örkened). är bosatta. Pilen anger familjernas ursprungsort 30 Fig. 16. Pedigrees över de tre svenska familjerna med hereditär pyrofosfatartropati. Röntgenfynd har markerats i den övre halvan och kliniska fynd i den nedre halvan av symbolerna för kvinna (cirkel) respektive m a n (fyrkant). Typiska fynd har markerats med svart och atypiska med streckad symbol. 31 Varierande debutålder och expressivitet Debutåldern i de tre familjerna var i genomsnitt 30 år, men några i familjen A och samtliga i C hade haft akuta artriter redan vid 20-22 års ålder. I övrigt var sjukdomsbilden och sjukdomens förlopp likartad i de tre familjerna. Framförallt var knän och andra stora leder engagerade, men symtom kunde påvisas även i perifera leder. Efter de första akuta artritanfallen övergick sjukdomen ofta i kroniska tillstånd något tiotal år senare. Särskilt i familj A var dessutom njursten och magsår vanligt förekommande. Det vanligaste röntgenologiska fyndet var diskreta förkalkningar i knäledens menisker. Nedärvningen synes vara autosomalt dominant med ofullständig penetrans och varierande expressivitet. Säker diagnos för ledförkalkning i två på varandra följande generationer kunde påvisas endast i två fall, båda mellan mor och dotter (11:8 och 111:39 i familj A samt 111:16 och IV: 14 i familj B). Nedärvning från såväl far som mor, till både söner och döttrar, är dock trolig med hänsyn till alla de olika fynden i de tre familjerna. Fig. 17. Genealogiska kopplingar mellan de tre familjerna med hereditär pyrofosfatartropati. Samtliga anfäder utom de som markerats med X var från Örkeneds församling i Kristianstads län. Anfäderna i generation I var födda åren 1700-1744. 32 Trolig genkälla i Kristianstads län De tre familjerna är besläktade, vilket framgår av fig. 17. Fam. A och C härstammar båda från par tre (första generationen), och C och B härstammar båda från kvinnan i par 8 och 9. Det finns dessutom släktskap utöver vad som framgår av figuren. Som exempel kan nämnas att sonen till par 12 bars fram i dopet av h u s t r u n i par 4, vilket enligt gammal svensk tradition tyder på nära släktrelationer. Stamfäder till familjerna A och C födda 1749 respektive 1756 var bröder. Den senare var gift med en syster till en stamfader i familj B. Samtliga tre familjer hade rötter bland anfäder inom samma församling (Örkened) i Kristianstads län (Fig. 17). Hur sjukdomsanlaget kom till Örkened vet vi inte, men med hänsyn till den starka koncentrationen till vissa områden i kontinentala Europa, är det sannolikt att någon därifrån kommit inflyttande med genen till södra Sverige (s.k. founder-effekt), kanske någon gång på 1600-talet. En teoretisk möjlighet är dock att det kan ha uppstått en nymutation i den aktuella sydsvenska populationen. En utförligare presentation av detta projekt har publicerats i Läkartidningen (26) och i Clinical Genetics (30). Resultaten har också presenterats i samband med medicinska riksstämman 1979 (se abstracts). 33 ORALA SPALTDEFEKTER De sjukdomar vi hittills berört är monogena, dvs det finns en speciell gen, som kan ge upphov till sjukdomen ifråga. Sådana monogena sjukdomar följer en mendelsk nedärvning och kan, som vi sett, vara autosomala eller X-kromosombundna, dominanta eller recessiva. Många ärftligt betingade sjukdomar (även egenskaper som hudfärg, kroppslängd m.m.) är dock polygena, vilket innebär att flera olika gener är involverade. Man kan se att sådana sjukdomar förekommer oftare i vissa familjer ä n i andra, men det går inte att påvisa något enkelt mendelskt nedärvningsmönster. Det är dessutom ofta både genetiska och miljömässiga faktorer som i samverkan ger upphov till olika sjukdomar och missbildningar. Man brukar i sådana fall tala om multifaktoriell etiologi. Orala slutningsdefekter, dvs gomspalter och läpp-käk-spalter med eller utan gomspalt, är typiska exempel på missbildningar som har sådan polygen och multifaktoriell orsaksbakgrund. Man kan säga att embryot är genetiskt programmerat, från befruktningsögonblicket, med gener som ger bättre eller sämre förutsättningar för vävnadsanlagen att växa ihop till sluten läpp, käke och/eller gom. Det kan uttiyckas så att det finns ett tröskelvärde under vilket den genetiska programmeringen är så ogynnsam att barnet under alla förutsättningar föds med en spalt i munhålan. Ovanför det tröskelvärdet kan vävnadsanlagen för läpp, käke, gom hinna växa ut fullständigt, men j u närmare tröskelvärdet, j u större är risken att ogynnsamma miljöfaktorer, t.ex.att mamman röker, skall ge upphov till en spaltdefekt. Epidemiologisk fall-referent-metodik I epidemiologiska studier använder man sig av kohort- eller fallreferentmetodik (50). Den förra innebär att man jämför exponerade populationer (kohorter) med referenser som anses vara mindre exponerade, för att se om det är fler som drabbats av skada eller sjukdom i de exponerade kohorterna. Den andra metoden innebär i stället att m a n jämför en grupp människor som drabbats av en sjukdom eller skada (fall) med en i övrigt jämförbar grupp friska individer (referenter) för att se om de drabbade utsatts för något speciellt agens, som kan ha gett upphov till sjukligheten. Fall-referentmetodiken är möjlig att använda även för att studera skillnader i genetisk påverkan, genom att jämföra antavlor bland fall med antavlor bland referenter. Många barn med läpp-käk-gomspalter i Skellefteåregionen Incidensen för såväl isolerad gomspalt som läppspalt med eller utan gomspalt tycks vara högre i Skellefteåregionen ä n i andra delar av Västerbottens län (tab. 1). En inventering av alla barn som fötts i Skellefteåregionen 19601980 bekräftade tidigare resultat, som presenterats av Beckman och Marie Nordström (1976). Av totalt 21.389 levande födda hade 29 (1,36 per tusen) gomspalt och 31 (1,45 per tusen) läppspalt med eller u t a n gomspalt. Incidensen för sådana spaltdefekter är lägre i såväl Umeå som Lycksele sjukhusregion. 34 T a b . l . Incidens för orala spaltdefekter bland levande födda. Sjukhusregion Gomspalt n °/oo Skellefteå (1) 29 30 21 1,36 1,27 0,72 8 0,98 Skellefteå (2) Umeå (2) Lycksele (2) Läppspalt Antal n ° / o o födda 31 34 28 7 1,45 1,44 0,96 0,86 21.389 23.614 29.261 8.186 (1) Nordström (1985), publ. 48 i publ.listan. (2) Beckman och Nordström, Marie (1976). Till vart och ett av de 60 barnen med spaltdefekt söktes ett referensbarn av samma kön, som hade fötts i samma församling och så nära i tiden som möjligt. Undersökningsområdet omfattar sex ursprungliga församlingar, vilka n u uppdelats i 14 mindre församlingar (fig. 18}. Fig. 18. Karta som visar de sex ursprungliga församlingarna (A-F) inom Skellefteåregionen samt de åtta församlingar som nybildats vid angivet årtal. 35 Med hjälp av resurser vid Demografiska databasens filial i Jörn spårades anfäder till de 60 spaltbarnen och de 60 referensbarnen åtta generationer bakåt i tiden. Antavlor för de fyra första generationerna registrerades med PC. Förhöjd frekvens spalter pga gener från norr Anfäder till spaltbarnen kom, jämfört med referensbarnens, i större utsträckning från Skellefteåregionen. Särskilt vanligt var det att spaltbarnen hade rötter i de tre nordliga urförsamlingarna (Norsjö, Jörn, Byske), vilket talar för ett geninflytande därifrån. En jämförelse av antavlorna visade att ingiften var vanligare bland spaltbarnens anfäder. Man har tidigare antagit att isolerad gomspalt och läppspalt med eller utan gomspalt representerar olika genetiska varianter av orala slutningsdefekter. Föreliggande resultat talar för att det finns speciella gener som ökar risken för båda dessa varianter, och att sådana gener förekommer rikligare i populationer från norra delen av Skellefteåregionen ä n i andra västerbottniska populationer. Sannolikt förekommer dock vissa "huvudgener" (major genes) som framförallt ökar risken för isolerad gomspalt, medan andra kanske främst ökar risken för läppspalter. För den som vill få dessa resultat närmare belysta med siffror hänvisas till en publikation i Hereditas (48). Projektet presenterades vid medicinska riksstämman 1983 (se abstracts). Ny metod - teoretiska tillämpningar De två antavlorna som visas ifig. 19 kan kanske göra det lättare att förstå principen för genealogisk fall-referentmetodik. Överst ser vi 32 teoretiskt möjliga anfäder till en viss proband fem generationer bakåt i tiden. I den nedre delen av bilden ser vi h u r det kan se ut i verkligheten. (I den aktuella studien spårade vi anfäderna ytterligare tre generationer bakåt.) Samma individ kan förekomma, inte bara i olika antavlor, utan i olika positioner inom samma antavla. Det verkliga antalet individer i generation I (fig. 19) har minskat från 32 teoretiskt möjliga till 17. Vi ser också att syskon (nr. 13 och 22) har gift in sig i olika generationer. Därför finns det ytterligare fyra anfäder inritade, vilka egentligen tillhör både generation I och generationen innan. Genom att ge alla anfäder ett kodnummer enligt den övre delen av fig. 19 kan man i ett anaregister direkt se var i en viss antavla en man (jämna nummer) eller kvinna (udda nummer) hör hemma. Föräldrarna till ana nr 32 blir alltså 64 och 65, farföräldrarna 128 och 129, morföräldrarna 130 och 131. Svärföräldrarna till 32, alltså föräldrarna till 33, blir 66 och 67, osv. Genom att välja referensbarn med motsvarande födelseår och födelseförsamlingar, som fallen, kan man få fram antavlor som bör spegla det för perioden och regionen (populationen) normala genealogiska mönstret. Antavlorna för fallen, i detta projekt barn med läpp-käk-gomspalter, kan genom graden av ingifte, och genom att anfäder i högre utsräckning kommer från en viss region (population), återspegla ett geninflytande via dessa anfäder. Om det inte finns speciella gener som ökar risken för en viss sjukdom eller missbildning, bör antavlorna för fallen och referenserna båda återspegla det för området normala genealogiska mönstret. 36 Fig. 19. Teoretiska antavlor som illustrerar principen för genealogisk fall-referéntmetodik. 37 Det finns många andra missbildningar, som har polygen och multifaktoriell orsaksbakgrund. Det är därför angeläget att genealogisk fall-referentmetodik kommer till användning i fler projekt i framtiden. Av särskilt intresse skulle det vara att använda sådan metodik i studier av neurala slutningsdefekter. Med sådana avses A. anencefali (acrania) - avsaknad av storhjärna B. encefalocele - hjärnbråck C. myelomeningocele - lyggmärgsbråck Även sådana missbildningar är vanligare i Skellefteåregionen än i andra delar av Västerbotten, vilket framgår av tab.2. Tab.2. Neurala slutningsdefekter bland barn födda 1965-1983. Sjukhus Skellefteå Umeå Lycksele* Total Barn med (se ovan) Per Antal 1000 födda A. B. C. A-C 9 7 18 10 0 1 2 2 4 28 19 6 0,6 0,7 19.019 31.509 8.777 16 5 32 53 0,9 59.305 * inkluderar Vilhelmina BB 1.5 39 BLODGRUPPEN "lilla p" Alldeles i böljan av 1900-talet kunde Landsteiner och hans lärjungar kartlägga AB0(noll)-systemet. Beroende på om den ena eller båda eller ingen av antigenerna A respektive B finns på de röda blodkropparnas yta tillhör man blodgruppen A, B, AB eller noll. Förekomsten eller avsaknaden av dessa antigener regleras genetiskt. Ett kvarts sekel senare upptäcktes blodgruppssystemen MN och P, där andra antigener är aktuella. Från 1940 och framåt har sedan en lång rad blodgruppssystem beskrivits. Bland dessa är Rhesusfaktom mest känd. Rhesus-positiva (Rh+) individer har, medan Rhesus-negativa (Rh-) saknar denna blodgruppsfaktor. Sällsynt överallt i världen utom i Västerbotten Blodgruppen "lilla p" tillhör P-systemet, som upptäcktes av Landsteiner och Levine 1927. Då kände man emellertid till endast två P-blodgrupper, P j och P 2 , vilka kontrolleras av olika gener (alleler) i ett visst genlokus (lokus = "läge", plur. loci). I mitten av 50talet föreslog Sånger att en tredje allel i detta genlokus kunde ge upphov till en blodgruppsfaktor, som några år tidigare beskrivits av Levine och medarbetare under benämningen Tj(a-). Idag är flertalet forskare överens om att det finns tre alternativa alleler som kan förekomma i P-lokus. Genen P j dominerar över P 2 och p, medan P 2 dominerar över p. Tab.3 visar samband mellan blodgrupper och olika genotyper för detta lokus. Tab.3. Samband mellan genotyper och fenotyper för P-systemet. GENOTYP p p ipi; p 2 2; pp p p ip2; 2P BLODGRUPP (fenotyp) p iP p l p 2 p ("lilla p") Blodgruppen lilla p är mycket sällsynt över hela världen. Man har beräknat att den normalt förekommer hos kanske en eller två av en million individer. Bertil Cedergren vid blodcentralen, regionsjukhuset, Umeå, visade emellertid i början av 70-talet, att denna sällsynta blodgrupp fanns hos åtta av 40.000 undersökta västerbottningar. Med utgångspunkt från detta material kunde h a n beräkna att p-blodgruppen är 70 gånger vanligare i Västerbotten än i andra delar av världen. Genfrekvensen (för p-genen) beräknades vara cirka en procent i Västerbotten som helhet. I samarbete med dr Cedergren har jag närmare analyserat pgenens förekomst och spridning i Västerbotten. Totalt 31 västerbottningar med blodgruppen "lilla p", fördelade på 20 familjer, var kända när studien inleddes. Föräldrarna i samtliga dessa familjer, med undantag av en där föräldrarna var födda söder om länsgränsen, kom från församlingar i östra delen av Västerbottens 40 län (fig. 20), där genfrekvensen därför kan beräknas vara två procent. Dr Cedergren hade, med hjälp av en sedermera avliden släktforskare, inlett vissa genealogiska utredningar. Dessa granskades och kompletterades med hjälp av personal vid Demografiska databasens filial i Jörn, varefter alla identifierade anfäder födda före 1875 registrerades i ett anaregister med födelsedata, namn och ort (i regel = födelseort) samt ett kodnummer som indikerar familj och position i antavlan. Registreringen genomfördes för att möjliggöra sortering av anorna efter familj, namn, födelsedata och boende/födelseort. Efter genomförd registrering erhöll samtliga inblandade en kopia av respektive familjs antavla. Fig.20. Karta som visar födelseorter för föräldrar till individer med blodgruppen "lilla p". 41 Genkälla i Skråmträsk En analys av anaregistret, som omfattar 7.184 poster, visade att ett stort antal anfäder härstammar från byar inom ett område kring Skellefteå i norra Västerbotten (inrutat ifig. 20). En man från byn Skråmträsk, född 1655, och hans hustru, född omkr 1660, var anfäder till 9 av de 20 familjer som omfattas av studien (fig.21). Någon av dessa anfäder tycks ha fört "lilla p"-genen vidare via sex av sina barn. Som framgår av fig.21 är föräldrarna i fam.4b kusiner (farfar och morfar är bröder). Fadern i 4b är bror till modern i 4a. Inget annat släktskap mellan familjerna i detta pedigree var känt före den genealogiska utredningen. Däremot framkom vid intervjuer med berörda familjer att de båda föräldrarna är kusiner även i fam. 10 och tremänningar i fam. 5, 6 och 12. Gemensamma anfäder till de båda föräldrarna kunde påvisas i 15 av de totalt 20 familjerna. Om en mer heltäckande genealogisk utredning hade varit möjlig, skulle sannolikt fler av familjerna kunna knytas till anfäderna i Skråmträsk (fig.21). De slutsatser vi kan dra, trots att det finns luckor i det genealogiska materialet, är att det fanns en genkälla för den recessiva pgenen i norra Västerbotten redan på 1600-talet, kanske ändå tidigare. Det ger en förklaring till att blodgruppen "lilla p" är så vanlig här jämfört med andra delar av Sverige och världen. Resultaten har publicerats i Human Heredity (58) och i samband med medicinska riksstämman 1988 (se abstracts). Fig.21. Pedigree som visar att nio västerbottniska familjer med blodgruppen "lilla p" kan knytas till gemensam anfader från mitten av 1600-talet. 43 TAPETORETINALA DEGENERATIONER Tapetoretinal degeneration kan användas som samlingsnamn för en rad degenerativa tillstånd i ögats näthinna. Dit hör retinitis pigmentosa (rp), som i sin tur representerar en heterogen grupp av sådana sjukdomar. Det är ofullständigt känt hur många ärftliga varianter som förekommer, men det rör sig säkert om minst något tiotal. Man har uppskattat att cirka 85 procent av fallen med ärftligt betingad rp i Sverige är autosomalt recessiva och 10 procent autosomalt dominanta, medan 5 procent är X-kromosombundna varianter. Sådan genetisk heterogenitet kan vara svår att upptäcka. Å andra sidan kan en och samma gen ge upphov till varierande sjukdomsbild. Så är, som vi sett ovan, fallet med hereditär maculadegeneration av typen Bests sjukdom, som är en form av central tapetoretinal degeneration. Tapetoretinala degenerationer, inklusive maculadegenerationer, kan dessutom orsakas av andra faktorer ä n genetiska. Det är vanligt att äldre människor drabbas av sådana ögonskador. Samtidigt kan i vissa fall såväl maculadegenerationer som rp vara ärftligt betingade även om debutåldern är genomgående hög. Den genetiska analysen är sålunda viktig, dels för att öka möjligheten att ställa rätt diagnos, dels för att berörda patienter och familjer ska kunna få så korrekt upplysning som möjligt om arvsrisker. Noggrann kartläggning av familjer med ärftliga sjukdomar är också nödvändiga för framtida möjligheter att gentekniskt lokalisera gener och närmare studera deras funktion, för att om möjligt finna bot mot dessa sjukdomar. Många varianter Typiska symtom vid autosomal recessiv rp är pigmentförskjutningar och pigmentklumpar i näthinnan, begränsning av synfältet och nattblindhet. Sjukdomen leder ofta till blindhet. I Västerbotten förekommer många delvis svårtydda varianter av sådan rp och liknande tapetoretinala degenerationer. En del mer eller mindre atypiska former har kallats 'Västerbottenchoroidit", men detta är ingen klar och entydig diagnos. I det projekt som här skall redovisas har vi sökt klarlägga om det kan finnas någon bestämd genkälla för tapetoretinala degenerationer i norra Sverige, och om i så fall gener från en sådan genkälla kan ge upphov till varierande sjukdomsbild eller till någon mer speciell variant av rp eller liknande tapetoretinal degeneration. Genom granskning av journalmaterial vid ögonklinikerna samt genomgång av register hos synkonsulenterna i Västerbottens län identifierades 166 patienter med någon form av sannolikt hereditär tapetoretinal degeneration. Ur detta grundmaterial gallrades alla som avlidit, hade mycket hög ålder eller som inte kunde återfinnas i länsstyrelsens adressregister (totalt 36 fall). Till återstående 130 skickades brev med information om projektet samt förfrågan om medgivande till genealogiska utredningar inkluderande registrering av anfäder i ett speciellt anaregister. En av de tillfrågade familjerna, med tre kända fall av rp, samt ytterligare tre sporadiska fall, ville inte delta i projektet, varför de uteslöts. Efter genomförd genealogisk utredning har vidare fyra familjer med dominant nedärvning (19 fall) samt en familj med två kända fall av rp och dövhet (sannolikt Ushers syndrom) tills vidare uteslutits ur materialet, som därefter omfattade 20 familjer med 50 44 kända fall samt 53 sporadiska fall av rp (inkl 8 fall av rp och dövhet) eller liknande tapetoretinal degeneration. Familjerna resp. de sporadiska fallen grupperades i någon av sex grupper efter den huvudsakliga diagnos de fått i journaler och/eller synkonsulenternas register. De sex grupperna är följande (tab.4): retinitis pigmentosa (rp), tapetoretinal degeneration (trd), retinitis punctata albescens (rpa), ehorioretinit eller retinoehoroidit (cr/rc), degeneratio retinae eller retinal degeneration (dr/rd) samt retinitis pigmentosa inkl. dövhet (rp+d). Tab.4. Familjer och sporadiska fall med tapetoretinala degenerationer (enligt diagnoser ovan). Diagnos Familjer Antal fam. Antal fall Sporad. fall Antal rp trd rpa cr/rc dr/rd rp+d F01-F06 F07-F15 F16-F20 6(2) 9(3) 5(2) 16 (5) 22(10) 12(5) Alla F01-F20 20 (7) 50(20) S01-S18 S19-S34 S35-S40 S41-S43 S44-S45 S47-S53 S01-S53 18(9) 16(5) 6(2) 3(1) 2(2) 8(2) 53(21) - - - - - - Utgående från en proband per familj och de 53 sporadiska fallen spårades föräldrarnas anor så långt bakåt som möjligt med hjälp av resurser vid Demografiska databasens central i Jörn. Alla identifierade anor med känt födelseår (födda före 1890) registrerades med PC (12.033 poster). Vaije ana (post) försågs med kodbeteckning som anger position i antavlan, om det är en ana på fars eller mors sida samt anknytning till respektive proband/fall. Vidare innehåller posterna uppgift om namn, födelsedatum samt födelseort (församling och by), så långt det var möjligt att erhålla uppgifter om detta. Genkälla i Burträskregionen Totalt kunde 7 av de 20 familjerna och 21 av de 53 sporadiska fallen knytas till anfäder i Burträskregionen födda på 1600-talet respektive 1700-talet (ftg.22). Som framgår av tab.4 var det fall med varierande diagnos som kunde knytas till dessa gemensamma anfäder. 1 1 7 fall kunde båda föräldrarna, i 7 fall endast far och i 4 fall endast mor knytas till någon av de aktuella anfäderna. B l och B2 (fig.22), födda 1654 respektive 1645, var med stor sannolikhet systrar. De hade samma efternamn och var födda i samma by inom församlingen. De gifte sig med två bröder från en by i Skellefteå landsförsamling. Ytterligare två anor i första generationen, födda 1651 (A) respektive 1666 (C), var från Burträsk. 45 Fig.22. Pedigrees som visar att 7 familjer och 21 sporadiska fall med tapetoretinal degeneration kan knytas till gemensamma anfäder i Burträsk (markerade med kryss). S33 finns med i båda dessa pedigrees, i det övre på sin fars och i det nedre på sin mors sida. 46 Anorna vid D l , D2 och E var alla födda inom Burträsk församling, men det var inte möjligt att spåra deras rötter längre tillbaka i tiden. I vissa fall kunde ett stort antal anfäder spåras. Det genealogiska materialet är emellertid ofullständigt. Luckor förekommer dels på grund av ofullständiga uppgifter i kyrkoarkivalierna (okänt födelseår, okänd far, okänd härstamning), dels på grund av att en del av de gamla kyrkböckerna skadats genom brand och därför är mycket svårtydda. I genomsnitt 221 anfäder per familj kunde spåras när vi funnit koppling till anfäderna i Burträskregionen jämfört med endast 122 bland övriga (tab.5). Ännu större var denna skillnad beträffande sporadiska fall, där i genomsnitt 292 anfäder spårats per fall, när de kunnat knytas till Burträsk, jämfört med endast 86 för övriga. Detta ger en antydan om att ännu fler fall av tapetoretinal degeneration i Västerbotten hade kunnat knytas till en sannolik genkälla i Burträskregionen om det genealogiska materialet varit mer fullständigt. Tab.5. Skillnader i antal spårade anfäder. Familjer/ sporadiska fall Familjer i pedigree övriga Sporadiska fall i pedigree övriga Antal Genomsnittligt antal spårade anfäder till far mor far+mor 7 13 109 49 112 73 221 122 21 32 124 41 168 45 292 86 Gemensam anfader med "lilla p" familjer Som nämnts ovan har vi kunnat knyta ungefär hälften av alla västerbottningar med blodgruppen "lilla p" till anfäder i byn Skråmträsk i Skellefteå landsförsamling. Dessa finns också med som anor till 4 av familjerna och 7 av de sporadiska fallen med tapetoretinal degeneration. Anledningen till detta visade sig vara att de båda bröderna vid B1/B2 (fig.22) kom j u s t från Skråmträsk, och dit flyttade de båda systrarna från Burträsk i samband med giftermålet. På så sätt kom ättlingar till rp-anorna att gifta sig med ättlingar till fyra av de sex barnen till p-anorna (fig.21). En viktig lärdom vi kan dra av detta är att man lätt kan hamna fel vid mer eller mindre osystematiska genealogiska utredningar. Hade vi inte förutsättningslöst spårat så många anfäder som möjligt, och sedan registrerat dem i ett anaregister som underlag för den fortsatta genealogiska analysen, hade vi lätt kunnat ledas in på ett sidospår, som t ex hade kunnat leda fram till p-anorna i Skellefteå landsförsamling i stället för till rp-anorna i Burträskregionen. 47 Det kan alltså vara lätt att hamna på sidospår vid sökandet efter genealogiska länkningar, och det finns luckor i det material man kan få fram ur kyrkoarkivalierna. Med den metodik som här använts, med registrering av alla identifierade anfäder, finns dock möjligheter att finna en möjlig genkälla för en specifik gen. Den slutsats vi kan dra från denna studie är sålunda att det sannolikt finns en autosomal recessiv gen i den västerbottniska populationen, som härstammar från en genkälla i Burträskregionen, och som kan ge upphov till rp och rp-liknande symtom med varierande sjukdomsbild. En förklaring till att två sporadiska fall med rp och dövhet (S 47 och S 53) kan knytas till anfäderna i Burträsk, kan vara att de har samma ärftliga typ av rp som de övriga fallen och att dövheten orsakats av andra faktorer, som inte behöver vara kopplade till synskadan. Resultat från detta projekt presenterades i samband med medicinska riksstämman 1990 (se abstracts) och är under publicering i vetenskaplig tidskrift (64). 49 PUBLIKATIONER 1. Nordström, S., Holmgren, G. and Thorburn, W.: A genetic study of congenital hereditary macular degeneration in the county of Västerbotten, Sweden. Acta OphthaL 50:539-542, 1972 2. Nordström, S., Holmgren, G. and Thorburn, W.: Hereditary macular degeneration in two pedigrees from northern Sweden. Human Heredity 22:86-90, 1972 3. Nordström, S.: Hereditary macular degeneration - A population survey in the county of Västerbotten, Sweden. Hereditas 78:41-62, 1974 4. Holmgren, G., Nordström, S. and Thorburn, W.: Urinary metabolic studies in hereditary macular degeneration. Acta OphthaL 52:225-230, 1974 5. Nordström, S.: Genealogical and genetical studies of hereditary macular degeneration in the county of Västerbotten, Sweden. Thesis, University of Umeå, Sweden, 1974 6. Nordström, S. and Barkman, Y.: Hereditary macular degeneration (HMD) in 246 cases traced to one gene source in central Sweden. Hereditas 84:163-176, 1976 7. Nordström, S.: Hereditär maculadegeneration i Västerbottens och Kopparbergs län. Läkartidningen 74:660-662, 1977 8. Nordström, S. och Johansson, E.: Husförhörens läsbetyg avslöjar ärftlig sjukdom. Forskning och Framsteg 1/1978, sid. 52-56 9. Björksten, B., Gustavsson, K-H., Eriksson, B., Lindholm, Å. and Nordström, S.: Chronie recurrent multifocal osteomyelitis and pustulosis palmo-plantaris. JPediatrics 93:227-231, 1978 10. Thorburn, W. and Nordström, S.: EOG in a large family with hereditary macular degeneration (Bests vitelliform macular dystrophy). Identification of gene carriers. Acta OphthaL 56:445-469, 1978 11. Nordström, S., Beckman, L. and Nordenson, I.: Occupational and environmental risks in and around a smelter in northern Sweden. I. Variations in birth weight. Hereditas 88:43-46, 1978 51 12. Nordenson, I., Beckman, G., Beckman, L. and Nordström, S.: Occupational and environmental risks in and around a smelter in northern Sweden. II. Chromosomal aberrations in workers exposed to arsenic. Hereditas 88:47-50, 1978 13. Nordström, S., Beckman, L. and Nordenson, I.: Occupational and environmental risks in and around a smelter in northern Sweden. III. Frequencies of spontaneous abortion. Hereditas 88:51-54, 1978 14. Nordenson, I., Beckman, G., Beckman, L. and Nordström, S.: Occupational and environmental risks in and around a smelter in northern Sweden. IV. Chromosomal aberrations in workers exposed to lead. Hereditas 88:263-267, 1978 15. Nordström, S., Beckman, L. and Nordenson, I.: Occupational and environmental risks in and around a smelter in northern Sweden. V. Spontaneous abortion among female employees and decreased birthweight in their offspring. Hereditas 90:291-296, 1979 16. Nordström, S., Beckman, L. and Nordenson, I.: Occupational and environmental risks in and around a smelter in northern Seden. VI. Congenital malformations. Hereditas 90:297-302, 1979 17. Beckman, G., Beckman, L. Nordenson, I. and Nordström, S.: Studies of spontaneous abortion, malformations and birthweight in populations exposed to environmental pollutants; Theoretical considerations and empirical data. In KBerg (Ed.): Genetic Damage in Man caused by Environmental Agents. Academic Press, New York, 1979 (pp 317-326) 18. Beckman, G., Beckman, L., Nordenson, I. and Nordström, S.: Chromosomal aberrations in workers exposed to arsenic. In KBerg (Ed.): Genetic Damage in Man caused by Environmental Agents. Academic Press, New York, 1979 (pp 205-211) 19. Nordström, S.: Genealogical tracing of genetical disorders in man; Application of parish catechetical meeting examination records (PCMER). In J.Sundin and E.Söderlund (Eds.): Time Space and Man. Essays in Microdemography. A&W, Stockholm, and Humanities Press, Atlantic Highlands, N.J., 1979 (pp 49-52) 20. Polland, W. and Nordström, S.: Eight cases of congenital achromatopsia with amblyopia in two pedigrees from northern Sweden. Acta Ophthal 57:653-664, 1979 53 21. Nordström, S. and Polland, W.: Different expressions of one gene for congenital achromatopsia with amblyopia in northern Sweden. Human Heredity 30:122-128, 1980 22. Nordström, S. and Thorburn, W.: Dominantly inherited macular degeneration (Bests disease) in a homozygous father with 11 children. CUnicaLGenetics 18:211-216, 1980 23. Nordström, S.: Epidemiology of hereditary retinal disorders in northern Sweden. Nordic CouncilArct. Med. Res. Rep. 26:55-57, 1980 24. Nordström, S.: Epidemiological studies of hereditary macular degeneration (Bests disease) in Swedish and Swedish-American populations. In A.W.Eriksson (Ed.): Population Structure and Genetic Disorders. Academic Press, London, 1980 (pp 431-443) 25. Nordström, S. and Beckman, L.: Variations in birthweight in areas with different degrees of urbanization and industrialization. Ambio 10:24-25, 1981 26. Bjelle, A., Hagstam, Å. and Nordström, S.: Hereditär pyrofosfatartropati i tre svenska familjer. Läkartidningen 78:4562-4566, 1981 27. Nordström, S.: Hälsa - vad är det? Biologen Nr 4, 1981, sid. 15-17 28. Beckman, L., Nordenson, I. and Nordström, S.: Occupational and environmental risks in and around a smelter in northern Sweden. VIII. Three-year follow-up of chromosomal aberrations in workers exposed to lead. Hereditas 96:261-264, 1982 29. Nordenson, I., Nordström, S., Sweins, A. and Beckman, L.: Chromosomal aberrations in lead exposed workers. Hereditas 96:265-268, 1982 30. Bjelle, A., Nordström, S. and Hagstam, Å.: Hereditary pyrophosphate artropathy (familial articular chondrocalcinosis) in Sweden. CUnical Genetics 21:174-180, 1982 31. Beckman, L. and Nordström, S.: Occupational and environmental risks in and around a smelter in northern Sweden. IX. Fetal mortality among wives of smelter workers. Hereditas 97:1-7, 1982 55 32. Hansson Mild, K., Lövstrand, K.G., Nordenson, I., Nordström, S. och Öberg, P.Å.: Biologiska effekter av lågfrekventa elektromagnetiska fält. Läkartidningen 79:1706-1710, 1982 33. Nordström, S.: Kyrkböcker avslöjar ärftliga sjukdomar. Forskning och Framsteg 7/1982, sid.39-43 34. Nordström, S.: Miljörisker för ofödda barn. Miljö i Sverige 2/1982, sid. 18-21 35. Nordström, S.: Vad är liv? (Debattinlägg) Forskning och Framsteg 4/1982, sid.48-49 36. Nordström, S. och Forsberg, B.: Missfallen i Teckomatorp - en fråga om redovisning av forskningsresultat. (Debattinlägg) Läkartidningen 79:4819-4820, 1982 37. Nordström, S., Birke, E. and Gustavsson, L.: Reproductive hazards among workers at high voltage substations. Bioelectromagnetics 4:91-101, 1983 38. Leffler, P. och Nordström, S.: Metaller i maternellt och fetalt blod. Undersökning av eventuella variationer i placentas barriärfunktion. Projekt Kol-Hålsa-Milfö, Teknisk rapport nr 71, 1983 39. Nordström, S. och Forsberg, B.: Reproduktionstoxikologiska effekter av luftföroreningar. Epidemiologiska studier. Litteraturöversikt. Projekt Kol-Hålsa-Miljö, Teknisk rapport nr 83, 1983 40. Nordström, S.: Missfallen i Teckomatorp - slutreplik. (Debattinlägg) Läkartidningen 80:113-114, 1983 41. Forsberg. B. och Nordström, S.: Slumpfaktorn stoppar forskare i Teckomatorp. (Debattinlägg) Miljöaktuellt 2/1983, sid. 14 42. Nordström, S. och Forsberg, B.: Farligt fixera sig vid signifikansvärden. (Debattinlägg) Läkartidningen 80:1786-1787, 1983 43. Nordenson, I., Hansson Mild, K., Nordström, S., Sweins, A. and Birke, E.: Clastogenic effects in h u m a n lymphocytes of power frequency electric fields: In vivo and in vitro studies. Radiat. Envirort Biophys. 23:191-202, 1984 44. Nordström, S.: Vi måste våga dra slutsatser. (Debattinlägg) Forskning och Framsteg 2/1984, sid. 59 57 45. Nordström, S.: När livet blir till - Om etisk beredskap infor den gentekniska framtiden. Kristendom och Skola 1/1984, sid. 20-21 46. Nordström, S.: Kreationism och evolutionism. Kristendom och Skola 4/1984, sid. 8-9 47. Forsberg, B. and Nordström, S.: Misscarriages around a herbicide manufactoring company in Sweden. Ambio 14:110-111, 1985 48. Nordström, S.: Genealogical studies in families with oral clefts and reference families. Hereditas 103:211-219,1985 49. Nordström, S.: Genetisk genealogisk forskning. Meddelande nr 2 från Demografiska 1985 50. Forsberg, B. och Nordström, S.: Vad är epidemiologi? Miljö och Hälsa 1/1985, sid.6-11 51. Nordström, S.: Livet - en gåta. EFS förlaget, Uppsala, databasen, Umeå univ. 1985 (137 sid.) 52. Nordström, S.: Genetic and reproductive hazards in people working at or living close to a Swedish non-ferrous smelter. Department of Public Health a n d Environmental Studies, Univ. of Umeå, Sweden, Report No 1, 1987 53. Nordström, S.: Genforskning på gott och ont. I "Forskningens konsekvenser för människa och samhälle", Nämndenför Diakoni och Samhällsansvar, NDS - Socialetiska rapporter 1987:2 54. Nordström, S.: Genetisk genealogisk forskning i Skellefteå-regionen. I "Forskningsdagarna i Skellefteå 12-13 september 1986", Föreningen för Skellefteåforskning, Rapport nr 1, 1987 55. Nordström, S., Nordenson, I. and Hansson Mild, K.: Genetic and reproductive hazards in high voltage substations. In L.E.Anderson, B.J.Kelman and R.J.Weigel (Eds.): Interaction ofBiological Systems with Static and ELF Electric and Magnettc Fields. Pacific Northwest Laboratory, Richland Wa, USA, CONF841041, 1987 59 56. Nordström, S. and Forsberg, B.: Reproductive hazards in areas with complex mixtures of air pollution. InR.H.Gray, E.KChess, P.J.Melltnger, R.G.Riley and D.L. Springer (Eds.): Health and Environmental Research on Complex Organic Mixtures. Pacific Northwest Laboratory, Richland Wa, USA, CONF-851027, 1987 57. Nordström, S.: Manipulerad produkt eller välsignad livsfrukt. - En bok om etik och biomedicinsk teknik. Verbum, Stockholm, 1988 (131 sid.) 58. Nordström, S. and Cedergren, B.: Genetic genealogical studies in 20 north Swedish families with the rare blood group p. Human Heredity 39:20-25, 1989 59. Nordström, S.: Om att tro sig veta och veta att man tror. Ars Medicina 1-2/1989, sid, 19-21 60. Nordström, S.: Vi måste ha kvar vördnaden för livet. Kyrknyckeln 3/1989, sid.6-7 61. Nordström, S.: Evolutionism eller kreationism? Vem eller vad ska man egentligen tro på? Vår Lösen 2-3/1990, sid.83-90 62. Nordström, S.: Debatten om 'Vårt ursprung".(Debattinlägg) Föreningen för populärvetenskap. Medlemsblad sid. 4-6 1-2/1990, 63. Nordström, S.: Charles Darwin - hädare eller hedersman? - En bok om evolutionism och kreationism samt livets ursprung och mening. Cordia, Uppsala, 1990 (170 sid.) 64. Nordström, S.: Variable expression of tapetoretinal degeneration derived from a gene source in northern Sweden. Manus JÖr Human Heredity, 1991 61 ABSTRACTS TILL VETENSKAPLIGA KONFERENSER Nordström, S., Holmgren, G. and Thorburn, W.: A genetical study on congenital hereditaiy macular degeneration in the county of Västerbotten. The Second International Symposium in Circumpolar Health, Finland, 1971. Abstracts ofPapers, p 43 Nordström, S., Holmgren, G. och Thorburn, W.: En genetisk studie av kongenital hereditär maculadegeneration i Västerbottens län. Läkaresällskapets riksstämma, Stockholm, 1971. Sammanfattningsbok, p 148 Nordström, S.: Hereditär maculadegeneration i Västerbottens län. Läkaresällskapets riksstämma, Stockholm, SanruTTanfattningsbok, p 129 1974. Nordström, S. och Barkman, Y.: Hereditär maculadegeneration; Kliniska och epidemiologiska aspekter. Läkaresällskapets riksstämma, Stockholm, 1976. Sv. Läkaresållsk. Handl, Band 85, Häfte 5, p 146 Nordström, S., Beckman, L. och Nordenson. I.: Abortfrekvens och födelsevikt i Rönnskärsområdet. Läkaresällskapets riksstämma, Stockholm, 1977. Sv. Läkaresållsk. Handl, Band 86, Häfte 4, p 195 Nordenson, I., Beckman, G., Beckman, L. och Nordström, S.: Kromosombrott hos arbetare exponerade för bly. Läkaresällskapets riksstämma, Stockholm, 1977. Sv. Läkaresällsk. Handl, Band 86, Häfte 4, p 195 Nordström, S. and Thorburn, W.: Early detection of hereditary macular degeneration (HMD). European Society of Human Genetics. Oslo Symposium, Clinical Genetics 13:131-132, 1978 1977. Thorburn, W. och Nordström, S.: Hereditär maculadegeneration (Bests vitelliforma dystrofi); Identifiering av friska anlagsbärare med EOG. Läkaresällskapets riksstämma, Stockholm, 1977. Sv. Läkaresällsk. Handl, Band 86, Häfte 4, p 436 Nordström, S.: Epidemiological studies of hereditaiy macular degeneration in Swedish and Swedish-American populations. Sigrid Juselius VII Symposium Mariehamn, Åland, 1978. Book of Abstracts, p 17 Nordström, S., Beckman, L. och Nordenson, I.: Abort, missbildningar och födelsevikt i graviditeter hos kvinnor arbetande vid Rönnskärsverken. Läkaresällskapets riksstämma, Stockholm, 1978. Sv. Läkaresällsk. Handl, Band 87, Häfte 3, p 182 Oulu, 63 Nordström, S., Beckman, G., Beckman, L. and Nordenson, I.: Unfavourable pregnancy outcome in people working in and living close to a smelter in northern Sweden. InternationaL Conference on Critical Current Issues in Environmental Health Hazards, Tel Aviv, Israel 1979. Book of Abstracts, p 70 Nordström, S., Beckman, G., Beckman, L. and Nordenson, I.: Reproductive hazards among people working in and living close to a smelter in northern Sweden. European Society of Human Genetics. Southampton Symposium, 1979. Cliniccd Genetics 17:79-80, 1980 Nordström, S. och Polland, W.: Komplett och inkomplett achromatopsia. Kliniska varianter av samma genetiska sjukdom? Läkaresällskapets riksstämma, Stockholm, 1979. Sv. Läkaresällsk Handl, Band 88, Häfte 3, p 171 Nordström, S. och Beckman, L.: Effekter på graviditetsutfallet av exponering av män i Rönnskärsmilj ön. Läkaresällskapets riksstämma, Stockholm, 1979. Sv. Läkaresällsk. Handl, Band 88, Häfte 3, p 172 Nordström, S. och Thorburn, W.: Sjukdomsdebut hos anlagsbärare för hereditär maculadegeneration (Bests sjukdom) identifierade med EOG. Läkaresällskapets riksstämma, Stockholm, 1980. Sv. Läkaresällsk. Handl, Band 89, Häfte 5, p 139 Nordström, S.: Miljörisker för ofödda barn. Arbetsmedicinska kontaktdagar, Arbetarskyddsstyrelsen, 1981.Sammanfattningsbok p 74-75 Umeå, Nordström, S., Bjelle, A. och Hagstam, Å.: Hereditär pyrofosfatartropati i Sverige. Läkaresällskapets riksstämma, Stockholm, 1981. Sv. Läkaresällsk. Handl, Band 90, Häfte 5, p 185 Beckman, L., Nordenson, I., Nordström, S. och Sweins, A.: Ger blyexposition kromosomskador? Läkaresällskapets riksstämma, Stockholm, 1981. Sv. Läkaresällsk. Handl, Band 90, Häfte 5, p 187 Hansson Mild, K. (moderator), Lövstrand, K.G., Lövsund, P., Nordenson, I., Nordström, S. och Öberg, P.Å.: Biologiska effekter av lågfrekventa elektromagnetiska fält. Läkaresällskapets riksstämma, Stockholm, 1981 (symposium). Sv. Läkaresällsk. Handl, Band 90, Häfte 5, p 8 65 Hansson Mild, K., Nordenson, I., Sweins, A. and Nordström, S.: Induction of chromosome aberrations in h u m a n lymphocytes in vitro by electrical discharges. (Poster) Bioelectromagnetics Society. Fourth Annual Scientific Angeles, Ca, USA, 1982. Book oj Abstracts, p 52 Session, Los Nordström, S. och Forsberg, B.: Variation i födelsevikt mellan områden med olika grad av luftföroreningar. Läkaresällskapets riksstämma, Stockholm, 1983. Sv. Läkaresällsk. Handl, Band 92, Häfte 8, p 217 Nordström, S.: Genealogiska studier i familjer med orala spaltdefekter och referensfamiljer. Läkaresällskapets riksstämma, Stockholm, 1983. Sv. Läkaresällsk. Handl, Band 92, Häfte 8, p 153 Nordström, S. och Forsberg. B.: Fler missfall ä n förväntat i Teckomatorp under slutet av 1970-talet. Läkaresällskapets riksstämma, Stockholm 1983. Sv. Läkaresällsk. Handl, Band 92, Häfte 8, p 153 Nordström, S., Nordenson, I. and Hansson Mild, K.: Genetic and reproductive hazards in high voltage substations. Twenty-third Hanford Life Sciencies Symposium, Richland, Oct 1984. Book of Abstracts, p 48 Nordström, S. and Forsberg, B.: Reproductive hazards in areas with complex mixtures of air pollution. Twenty-fourth Hanjord Life Sciences Symposium, Richland, Oct 1985.Book of Abstracts, p 47 Nordström, S. och Cedergren, B.: Genkälla för blodgruppen "lilla p" spårad till norra Västerbotten. Läkaresällskapets riksstämma, Stockholm, 1988. Sv. Läkaresällsk. Handl, Band 97, Häfte 1, p 222 Nordström, S.: Varianter av retinitis pigmentosa orsakade av samma gen? Läkaresällskapets riksstämma, Stockholm, 1990. Sv. Läkaresällsk. Handl, Band 99, Häfte 1, p 207 Wa, USA, Wa, USA, 67 ÖVRIGA REFERENSER Andersson, R.: Familial amyloidosis with polyneuropathy. A clinieal study based on patients living in northern Sweden. ActaMed. Scand. SupplNo590, 1976 Barkman, Y.: A clinical study of a central tapetoretinal degeneration. Acta Ophthal 39:663-671, 1961 Beckman, L.: Arv och arvsrisker hos människan. Prisma, Stockholm, 1983 Beckman, L. and Bergenholtz, A.: Epidermolysis bullosa hereditaria letalis in northern Sweden. Hereditas 80:173-176, 1975 Beckman, L. and Nordström, M.: Population studies in northern Sweden. VIII. Frequencies of congenital malformations by region, time, sex and maternal age. Hereditas 84:35-40, 1976 Böök, J.A., Wetterberg, L. and Modrzewska, K.: Epidemiological and genetic investigations in a north Swedish geographical isolate. In A.W.Eriksson (Ed.): Population Structure and Genetic Disorders. Academic Press, London, 1980 pp 391-404 Drugge, U.: Om husförhörslängder som medicinsk urkund. Psykisk sjukdom och förståndshandikapp i en historisk källa. Scriptum Nr 8, Forskningsarkivet, Umeäuniv., 1988 Drugge, U., K:son Blomquist, H., Gustavson, K-H. and Holmgren, G.: Fragile X families in a northern Swedish county: A genealogical study of possible affected individuals in the nineteenth centuiy. Am J Med Gen, 1990 (In press) Hillborg, P. och Sundström, J.: Morbus Gaucher i Sverige. Utbredning och arvsförhållanden. Föredrag vid Genealogiska Föreningens årsmöte, Stockholm, 1979 23 nov. Holmgren, G., K:son Blomquist, H., Drugge, U. and Gustavson, KH.: Fragile X families in a northern Swedish county - A genealogical study demonstrating apparent paternal transmission from the 18th centuiy. AmJMed Gen 30:673-679, 1988 Jageli, S.: Sjögren Larsson Syndrome in Sweden. An epidemiological, genetic, clinical and biochemical study. Umeå university Medical Dissertations. New Series No 68, 1981 Lindsten, J . och Iselius, I. (Red.): Klinisk genetik. Natur & Kultur, Stockholm, 1987 69 McKusick, V.A.: Mendelian inheritance in man. Catalogs of autosomal dominant, autosomal recessive, and X-linked phenotypes. The Johns Hopkins University Press, Baltimore and London, Ninth Editton, 1990 Pettersson, O.P.: Gamla byar i Vilhelmina. Etnologiska källskrifter 1, Stockholm, 1941 Wetterberg, L.: A neuro-psychiatric and genetical investigation of acute intermittent porphyria. Svenska Bokförlaget (Norstedts), 1967 SCRIPTUM 1. Egil Johansson: BOKSTÄVERNAS INTÅG. Artiklar i folkundervisningens historia. I. 28 s. 1988. Pris: 20 kr. 2. Daniel Lindmark: BARNAUNDERVISNINGEN I HÄRNÖSANDS STIFT speglad i uppfostringskommitténs enkät 1813. 86 s. 1988. Pris: 40 kr. 3. Sten Henrysson: LAPP ELLER NYBYGGARE? I l s . 1988. Pris: 20 kr. 4. Karin Snellman: Förteckning över ERIK NORDBERGS ARKIV. 39 s. 1988. Pris: 30 kr. 5. Sten Henrysson: "LÄSTE LUTHERI CATECHES PÅ LAPSKA". Om religions- och läsundervisning i Jokkmokks socken före folkskolans införande. 31 s.1988. Pris: 30 kr. 6. Tuuli Forsgren: SAMISK KYRKO- OCH UNDERVISNINGSLITTERATUR I SVERIGE 1619-1850. 2 uppl. 66 s. 1988. Pris: 40 kr. 7. Egil Johansson: KUNSKAPENS TRÄD. Artiklar i folkundervisningens historia. II. 73 s. 1988. Pris: 40 kr. 8. Ulf Drugge: OM HUSFÖRHÖRSLÄNGDER SOM MEDICINSK URKUND. Psykisk sjukdom och förståndshandikapp i en historisk källa. 40 s. 1988. Pris: 40 kr. 9. "CATALOGUS DISCENTIUM vid Jockmocks Schola, ifrån Åhr 1732. tå hon tog sin böljan." En bearbetad och kompletterad elevmatrikel över Jokkmokks lappskola. Utg. av Anita Alm, Tuuli Forsgren & Sten Henrysson. 23 s. 1989. Pris: 20 kr. 10. Egil Johansson: LÄSER OCH FÖRSTÅR. Artiklar i folkundervisningens historia. III. 39 s. 1989. Pris: 40 kr. 11. Sölve Anderzén: LAPPMARKEN I LITTERATUREN. Valda studier inom ett forskningsprojekt. 153 s. 1989. Pris: 50 kr. 12. Greger Fröjd & Robert Olsson: LAPPMARKENS PRÄSTER 1593-1904. En studie av bakgrund och karriär. 42 s. 1989. Pris: 40 kr. 13. Margareta Attius Sohlman: ABC-BOKEN PÅ KYRKSLAVISKA. Bokstävernas vandring i österled. 36 s. 1989. Pris: 30 kr. 14. Sten Henrysson: JOKKMOKKS LAPPSKOLAS ELEVER 1732-1846. En analys. 26 s. 1989. Pris: 20 kr. 15. Sölve Anderzén:"... FÖRA DEM TIL BÄTTRE LIUS I CHRISTENDOMEN..." Undervisningen vid Jukkasjärvi skola och i Jukkasjärvi församling åren 1744-1820. 153 s. 1989. Pris: 50 kr. 16. Daniel Lindmark & Stephanus Neib: UNDERVISNINGEN I LAPPMARKEN enligt svaren på 1812 års uppfostringskommittés enkät. 47 s. 1989. Pris: 40 kr. 17. Sam Engman: FÖLLINGE LAPPSKOLA 1748 - 1818. 26 s. 1989. Pris: 30 kr. 18. ARJEPLOGS LAPPSKOLA. Bearbetade och kompletterade elevmatriklar omfattande åren 1743 -1820 och 1863 - 1875. Utg. av Carl-Henry Johansson & Johnny Flodin. 29 s. 1989. Pris: 30 kr. 19. Sten Henrysson: PRÄSTERNA I LAPPMARKEN FÖRE 1850. Ursprung och arbetsuppgifter. 27 s. 1989. Pris: 30 kr. 20. Daniel Lindmark: EN SKOLA FÖR STADEN, REGIONEN OCH KYRKAN. Elever, lärare och präster i Piteå skola före 1850. 133 s. 1990. Pris: 50 kr. 21. Carl-Henry Johansson & Johnny Flodin: ELEVERNA VID ARJEPLOGS LAPPSKOLA 1743 - 1820. En analys. 24 s. 1990. Pris: 30 kr. 22. Sven Lundkvist: DEN ÄLDRE SVENSKA FOLKBOKFÖRINGEN. - Harry Lenhammar: HISTORISK OCH KYRKOHISTORISK FORSKNING I REGIONALT PERSPEKTIV. Två föredrag vid en stiftshistorisk dag i Umeå den 26 oktober 1989. 20 s. 1990. Pris: 40 kr. 23. PRÄSTERNA OCH LIVET I LAPPMARKEN. Del 1. Sten Henrysson: Präster och skolmästare i Jokkmokks socken 1607-1850. Biografiska uppgifter. Del 2. Carl-Henry Johansson: Släktskap och ingiften. Några exempel. 31 s. 1990. Pris: 40 kr. 24. Sölve Anderzén: JUCKASJERFWI SCHOUE MATRICKEL. Inrättadt wid Scholans begynnelse år 1744. En rekonstruktion. 13 + XXI s. 1990. Pris: 40 kr. 25. Sölve Anderzén: ELEVERNA VID JUKKASJÄRVI LAPPSKOLA 1744-1820. En första analys. 30 s. 1990. Pris: 40 kr. 26. Tuuli Forsgren: "...FÖRST AT INHÄMTA SPRÅKET, OCH SEDAN DERUPPA LÄRA SIN CHRISTENDOM..." Om finska böcker och sameundervisning i Torne och Kemi lappmarker före 1850. 45 s. 1990. Pris: 50 kr. 27. Carl F. Hallencreutz: PEHR HÖGSTRÖM OCH ANNA OLOFSDOTTER. Ett bidrag till 1740-talets samiska kyrkohistoria. 15 s. 1990. Pris: 40 kr. 28. Daniel Lindmark: LÄS- OCH SKRIVKUNNIGHETEN FÖRE FOLKSKOLAN. Historisk läskunnighetsforskning i nordiskt och internationellt perspektiv. 43 s. 1990. Pris: 50 kr. 29. Stefan Nordström: GENEALOGI OCH GENETIK. Beskrivning av sex projektområden där kyrkböcker använts i genetisk forskning. 69 s. 1991. Pris: 50 kr. 30. ÅSELE LAPPSKOLA 1732-1820. Bearbetad och kompletterad elevmatrikel. Utg. av Carl-Henry Johansson & Johnny Flodin. 33 s. 1991. Pris: 40 kr. 31. Anita Alm & Sten Henrysson: GÄLLIVARE LAPPSKOLA 1756-1850. En elevmatrikel jämte analys. 36 s. 1991. Pris: 40 kr. 32. Bengt Hjalmar Andersson: ETT BIDRAG TILL HISTORIEN OM BYN SÄTTER/ JARÄMAI LULE LAPPMARK. 39 s. 1992. Pris: 50 kr. 33. Nils Eriksson: SAMESKOLOR INOM ÅSELE LAPPMARK. Utg. av Sten Henrysson. 373 s. 1992. Pris: 75 kr. 34. Margareta Attius Sohlman: BOKSTAVENS MAKT ÖVER SLAVISKA SJÄLAR. Uppsatser och artiklar i ämnen rörande projektet "Alphabeta varia. Tidiga ABCböcker i skilda kyrkotraditioner". 104 s. 1992. Pris: 60 kr. 35. Sten Henrysson & Johnny Flodin: SAMERNAS SKOLGÅNG TILL 1956. 82 s. 1992. Pris: 60 kr. 36. Sten Henrysson & Johnny Flodin: SAMERNAS SKOLGÅNG: 1957 ÅRS NOMADSKOLEUTREDNING. 29 s. 1992. Pris: 50 kr. 37. Sten Henrysson: DARWIN, RAS OCH NOMADSKOLA. Motiv till kåtaskolreformen 1913. 22 s. 1993. Pris: 50 kr. 38. Egil Johansson: KAN SJÄLVA ORDEN. Artiklar i folkundervisningens historia. IV. 67 s. 1993. Pris: 60 kr. 39. Sölve Anderzén:"...ATT LÄRA THEM SOM BEHÖFWA..." Undervisning, byabön och dop i Kemi och Torne lappmarker. Två bidrag till 1700-talets kyrkohistoria på Nordkalotten. 31 s. 1995. Pris: 50 kr. 40. Gennadij Kovalenko: SVERIGE OCH RYSSLAND UNDER 1600-TALET. Några episoder ur det politiska och kulturella livet. (Rysk text med korta resuméer på svenska). 41 s. 1995. Pris: 50 kr. 41. Sven Hansson: BYABÖN OCH BÖNBYAR I GAMLA UME SOCKEN. (Umeå, Sävar, Vännäs och Vindelns socknar). 106 s. 1996. Pris: 60 kr. 42. Sölve Anderzén: TEACHING AND CHURCH TRADITION IN THE KEMI AND TORNE LAPLANDS, NORTHERN SCANDINAVIA, IN THE 1700s. 33 s. 1996. Pris: 50 kr. [Översättning till engelska av Scriptum 39.] 43. Ingegerd Stenström: Förteckning över FAMILJEN LINDERS ARKIV. 53 s. 1996. Pris: 60 kr. 44. Mari Ericsson: "ETT ÄDELT KALL BLEV DIG GIVET." Småskollärarinnorna i Jukkasjärvi skoldistrikt 1912-1930. 48 s. 1998. Pris: 50 kr. 45. ISKYRKAN I JUKKASJÄRVI. Sakral symbol och pastoral funktion. Forskningsseminariet Turisten i Iskyrkan 21-23 februari 1997. Red. av Sölve Anderzén. 81 s. 1998. Pris: 100 kr. 46. Karin Snellman: Förteckning över HELMER GRUNDSTRÖMS ARKIV. 40 s. 1998. Pris: 60 kr. 47. Simone Pusch: NOMADSKOLINSPEKTÖRERNA OCH SOCIALDARWINISMEN 1917-1945. 42 s. 1998. Pris: 50 kr. 48. Edith M. Eriksson: HELMER OSSLUND OCH HANS SYSKON. Tidigare okänd korrespondens i familjearkiven. 57 s. 1999. Pris: 80 kr.