U - IFM
advertisement

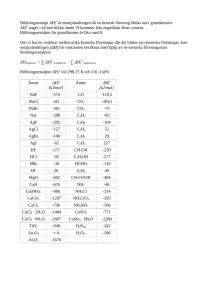
Kap. 7+8. Kemisk Termodynamik 7.1 Första huvudsatsen Termodynamik: ”Värmets rörelser”, läran om energins former och omvandlingar Energi: Storhet som ”medför förmåga att uträtta arbete” Arbete (w): w = − ∫ F dx eller dw = − F dx (Process som leder till rörelse mot motriktad kraft) Ex: pext = konst, Vf >Vi ⇒ w = -pext ⋅ (Vf – Vi) = -pext ⋅∆V ∆V > 0 ⇒ w < 0 Systemet utför arbete på omgivningen ∆V < 0 ⇒ w > 0 Omgivningen utför arbete på systemet Termodynamiskt tillstånd: Tillstånd som definieras av värdena på dess variabler såsom p, V och T. Inre energi (U): U = Epot + Ekin D.v.s. inre energin U för ett system av molekyler är summan av molekylernas lägesenergier och rörelseenergier. F’ = -F F x Vi har speciellt Tryck-volymsarbete: Termisk energi: kinetiska energin för partiklarnas slumpmässiga rörelser Temperatur kan definieras som ett mått på den genomsnittliga (slumpmässiga) kinetiska energin, exempelvis för ett system av partiklar: 3 m ⋅ v2 kB T = 2 2 Värme (q): Flöde av termisk energi Vf w = − ∫ pext dV Vi syst. omg. q q q > 0 : från omg. till system: endoterm adiabatisk process: process där q = 0 isotermisk process: process där T = konstant q < 0 : från system till omg.: exoterm Termodynamikens första lag eller 1:a Huvudsatsen: ∆U = w + q Alternativt uttryckt: Energin bevaras U är en så kallad tillståndsfunktion: dess värde beror bara av värdena på dess variabler såsom p,V,T. Lite fler olika definitioner: system: det vi mäter på omgivning: resten av universum Termodynamisk jämvikt: systemet sägs befinna sig i ett jämviktstillstånd ifall om systemet skulle frikopplas från omgivningen så ändras inte egenskaperna (exv U, T, p etc.) isolerat system: kan inte utbyta massa eller energi med omgivningen (⇒ q = 0, w = 0, ∆U = 0) ”termiskt isolerat”: kan inte utbyta värme med omgivningen, q = 0 Anm: För en ideal gas är Epot = 0 ⇒ U = Ekin ∝ T ⇒ Om isotermisk process så är ∆U = 0 (ty Uf = Ui om Tf = Ti och ∆U = Uf – Ui ) diatermisk process: process där Tsystem = Tomgivning (vanligen är Tomgivning = konst. ⇒ isotermisk process), denna process tillåter att värme överförs. Konstant-p process: p = konstant Konstant-V process: V = konstant (⇒ w = 0 ) Reversibel process: En process där en infinitisimal (= mycket liten) ändring av (en av) systemets parametrar kan ändra riktning på processen. Sker under jämviktsförhållanden. Ex: Reversibel expansion av ideal gas från Vi till Vf i en isotermisk process (T = konst) där: pext = p (gasens eget tryck, som varierar när V ändras) Betrakta en process där p = pext = konst. (”konstant-p-process”): w = – p ∆V q = qp (subskriptet betyder att p =konst) ⇒ ∆U = q + w = qp – p ∆V ⇒ qp = ∆U + p ∆V Anm: V = konstant: (w = 0 ⇒ ) qV = ∆U Värmeflöde till systemet kan orsaka en temperaturökning ∆T: q = C ∆T C = värmekapaciteten (olika för olika ämnen) Inför en ny storhet, entalpi H, som definieras: H = U + pV Vitsen: om konstant-p-process så är: ∆H = ∆U +∆(pV) = ∆U + p ∆V ⇒ 7.4 Termokemi Termokemi: Studiet av omsatt värme vid kemiska reaktioner qp = ∆H d.v.s. entalpiändringen är den värme som åtgår eller frigörs för en kemisk reaktion vid konstant tryck. Anm: Kemisk reaktion där n mol gas bildas (netto): ∆H = ∆U +∆(pV) = = ∆U + ∆(nRT) ⇒ ∆H = ∆U +∆n ·RT Kemisk reaktion: ”atomer i tillstånd (i)” → ”atomer i tillstånd (k)” Hi Hk ∆H = Hk - Hi Alternativt uttryckt, om vi har reaktionen: reaktanter → produkter så är reaktionsentalpin ( ∆H eller ∆Hrxn eller ∆rH ): ∆rH = H(produkter) – H(reaktanter) där H(produkter) är summan av produkternas entalpier etc. Standardtillstånd: Tillståndet för det rena ämnet vid trycket 1 bar och en viss temperatur (vanligen 298.15 K) Standardentalpi: Entalpin för standardtillståndet H°° Standardbildningsentalpi: Entalpiskillnaden för bildning av rena ämnen i deras stabila tillstånd vid po = 1 bar och temperaturen ifråga relativt grundämnena i deras standardtillstånd. Beteckning ∆fH°° eller ∆Hf° H2(g,p°) + ½ O2(g,p°) → H2O(l) Anm: Joner i vattenlösningar: ∆ f H o (H+ , aq) = 0 ) Dessutom gäller Hess lag: En reaktions ∆H kan skrivas som en summa av möjliga delreaktioners ∆H. Ex: ∆H° för C(s) + O2(g) → CO2(g) ? C(s) + ½O(g) → CO(g) ∆H°1 = -111 kJ/mol CO(g) + ½ O2(g) → CO2(g) ∆H°2 = -283 kJ/mol C(s) + O2(g) → CO2(g) Standardreaktionsentalpi ∆rH : o reaktanter (R) → produkter (P) n1 R1 + n2 R2 + … → m1 P1 + m2 P2 + … ∆r H o = produkter ∑m ⋅∆ i i f H (Pi ) − o m reaktanter ∑n ⋅∆ i f H mo (R i ) i ( ∆H m = entalpiändring per mol ) ( Några speciella beteckningar för olika reaktionsentalpier: ∆ trs H , ∆ fus H , ∆ vap H , ∆ sub H , ∆ hyd H , ∆ c H ∆H°=∆H°1+∆H°2 = -394 kJ/mol 8.1-2 Förlopps riktning • Betrakta två system med olika T som förs ihop till ett system med diatermisk skiljevägg: TA TB → → q TA > TB T T Samma temp Däremot observeras inte: T T → TA TB → TA TB TA > TB 1:a HS säger ingenting om processens riktning, bara att energin bevaras • Betrakta gasexpansion: → ← • Studsboll ∆H > 0 • Ba(OH)2(s)+2NH4NO3(s) → Ba(NO3)2(s)+2H2O(l) + 2NH3(aq) Spontan! VI behöver en till termodynamisk lag, ”2:a huvudsatsen”, som ska svara på frågan: Vad sker spontant? Oordning gynnas på ordningens bekostnad Dela upp Universum i ett lokalt system (som vi strax kort och gott skall kalla för ”systemet”) och omgivningen: omgivningen systemet Enligt 1:a HS är U = konstant för totala systemet (om universum är isolerat!) Entropi = storhet som kvantifierar 2:a HS. Beteckning: S 2:a Huvudsatsen: För entropi gäller I. ∆Suniv > 0 för spontan process II. ∆Suniv = 0 för reversibel process III. S (för lokala systemet) är en tillståndsfunktion där Suniv = S + Somgivning (S = Slokal) S kan relateras till mätbara storheter via två olika vägar: 1. Statistik (Boltzmann 1896) S = kB ln(W) W = antalet sätt på vilket tillståndet kan realiseras 2. Energetik (Termodynamisk definition) 8.3 Termodynamikens tredje lag Definition av entropi S för systemet: 3:e Huvudsatsen: För alla ämnen som är perfekta kristaller är S = 0 vid T = 0. dq reversibel dS = T Om T konstant under hela processen: ∆S = Motivering: S > 0 vid högre T och/eller icke-perfekta kristaller qreversibel T Tillfört värme ökar oordningen Lägre temp. medför större ökning för samma tillförda värmemängd 3:e HS ⇒ Absoluta värden på S(T) kan erhållas ⇒ Standardentropier S°(T) (rent ämne vid 1 bar) För omgivningen gäller: dSomgiv = dqomgiv Tomgiv där dqomgiv = − dq för den process som genomförs. För en spontan process (där T = Tomgiv): dS univ = dS + dS omgiv > 0 ⇔ dS univ = dS + dq omgiv T dS univ = dS + − dq T dS > dq T >0 ⇔ >0 ⇔ Clausius olikhet ⇒ Standardreaktionsentropier: ∆r S o = produkter ∑m ⋅S i i o (Pi ) − reaktanter ∑n ⋅S i i o (R i ) En fysikalisk innebörd hos fri energi: 8.4 Fri energi Antag system i termisk jämvikt med omgivningen vid T = konst, p = konst ⇒ dq = dH dq T dH ⇒ dS ≥ ⇔ dH − TdS ≤ 0 (∗) T Clausius olikhet: dS ≥ Definiera Gibbs Fria Energi: G = H − TS Infinitisimal förändring (T=konst, p=konst): dG = dH − TdS (∗) ⇒ dG ≤ 0 för spontan (dG < 0) eller reversibel/j.v. (dG = 0) process wadd,max = ∆G där wadd,max är det maximala arbetet utöver pVarbetet (p=konst). Standardbildnings-Gibbs fria energier ∆fG° : = ∆G relativt rena grundämnena i deras stabila faser vid trycket 1 bar Reaktioner: ∆rG° (eller ∆G°rxn ) ∆rG° = ∆rH° − T ∆rS° ∆rG° så stort och negativt som möjligt om: ∆rH° stort och negativt (exoterm) ∆rS° stort och positivt (oordningen i systemet ökar) För ”fullständiga” spontana (<) eller reversibla (=) processer: ∆G ≤ 0 ∆rG° kan vara negativt (d.v.s. spontan process) även om ∆rH° är positivt (endoterm) om ∆rS° är positivt och T är tillräckligt högt. ∆rG° kan vara negativt (d.v.s. spontan process) även om ∆rS° är negativt (oordningen minskar i systemet) om ∆rH° är negativt (exoterm) och T är lågt. 10.3 ∆rG°:s relation till jämv. konst. K Betrakta exempelvis gasfasreaktionen: b B (g) + c C (g) → d D (g) + e E(g) Om man vill ha ∆rG för andra (partial)tryck än standardtrycket 1 bar (såsom i en blandning): ∆ r G = ∆ r G° + RT ln Q där pDd pEe Q= b c pB pC Vid jämvikt (V) är ∆rG = 0 och Q = K 0 = ∆ r G° + RT ln K ⇒ ∆ r G° = − RT ln K ⇒