Cefotaxim Stragen powder for solution for injection or infusion SmPC
advertisement

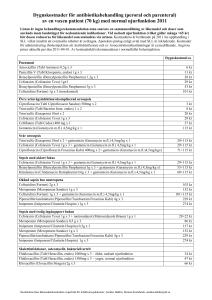
PRODUKTRESUMÉ 1 LÄKEMEDLETS NAMN Cefotaxim Stragen 0,5 g pulver till injektionsvätska, lösning Cefotaxim Stragen 1 g pulver till injektions- och infusionsvätska, lösning Cefotaxim Stragen 2 g pulver till injektions- och infusionsvätska, lösning 2 KVALITATIV OCH KVANTITATIV SAMMANSÄTTNING En injektionsflaska innehåller 0,5 g, 1 g eller 2 g cefotaxim (som cefotaximnatrium). För fullständig förteckning över hjälpämnen, se avsnitt 6.1 3 LÄKEMEDELSFORM 0,5 g: 1 och 2 g: Pulver till injektionsvätska, lösning. Pulver till injektions- och infusionsvätska, lösning Beskrivning: kristallint, vitt till svagt gulaktigt pulver. 4 KLINISKA UPPGIFTER 4.1 Terapeutiska indikationer Cefotaxim Stragen är indicerat för behandling av följande allvarliga infektioner, när man vet eller anser att det är troligt att infektionen orsakats av cefotaximkänsliga bakterier (se 5.1): - Bakteriell pneumoni (se avsnitt 4.4). - Allvarliga njurinfektioner och övre urinvägsinfektioner. - Svåra hud- och mjukdelsinfektioner. - Intraabdominella infektioner (inklusive peritonit). Vid intraabdominella infektioner bör cefotaxim kombineras med antibiotika med effekt på anaeroba bakterier. - Akut bakteriell meningit (speciellt när det orsakats av H. influenzae, N. meningitis, E. coli och Klebsiella stammar). - Sepsis Officiella riktlinjer för val av antibiotikum ska beaktas. Bakteriologiska tester och resistensbestämningar ska utföras. 4.2 Dosering och administreringssätt Cefotaxim Stragen kan efter beredning av lösningen, administreras som intravenös bolusinjektion, som intravenös infusion eller som intramuskulär injektion. Följ nedanstående instruktioner (se också 6.6). Intramuskulär injektion reserveras för exceptionella kliniska situationer, risk och nytta bör noga övervägas före användning. Speciella råd för intramuskulär injektion bör beaktas. Om lidokain används vid beredningen av lösningen för intramuskulärt bruk, bör hänsyn tas till produktinformationen för det lidokaininnehållande läkemedlet, speciellt med tanke på kontraindikationerna. Dosering och administreringssätt bestäms av infektionens svårighetsgrad, bakteriernas känslighet och patientens status. Behandlingen kan påbörjas innan resultaten av sensibilitetstesterna är kända. Cefotaxim och aminoglykosider har synergistiska effekter. Vuxna och ungdomar (från 12 år): Normaldosen till vuxna och ungdomar är 2-6 gram per dygn. Dygnsdosen bör delas upp på 2 doser var 12:e timme. Dosen kan dock varieras beroende på infektionens svårighetsgrad, bakteriens känslighet och patientens tillstånd. Doseringsrekommendationer: Vanliga infektioner vid närvaro av (eller misstanke därom) en känslig mikroorganism: 1 g var 12:e timme vilket motsvarar totalt 2 g per dygn. Infektioner vid närvaro av (eller misstanke om) flera känsliga eller måttligt känsliga mikroorganismer: 1-2 g var 12:e timme vilket motsvarar en total dygnsdos på 2-4 g. Allvarliga infektioner orsakade av oidentifierade mikroorganismer eller infektioner som inte kan lokaliseras: 2-3 g per doseringstillfälle 3-4 gånger per dygn var 8:e -6:e timma, maximalt 12 g per dygn. Spädbarn och små barn (28 dagar till 23 månader) och barn (2 till 11 år): Normaldos till spädbarn, småbarn och barn < 50 kg är 50-150 mg/kg/dygn uppdelat på 2-4 doser. Vid mycket allvarliga infektioner kan upp till 200 mg/kg/dygn krävas uppdelat på flera doser. Till barn > 50 kg ges normal vuxendos men den maximala dygnsdosen 12 g ska inte överstigas. Nyfödda (0-27 dagar): Den rekommenderade dosen är 50 mg/kg/dygn uppdelat på 2-4 doser (var 12:e till 6:e timme). Vid livshotande infektioner kan det vara nödvändigt att öka den dagliga dosen till 150-200 mg/kg/dygn, hänsyn måste tas till skillnader i njurarnas och njurfunktionens utveckling (se även särskilda doseringsrekommendationer vid meningit). För tidigt födda barn: Den rekommenderade dosen är 50 mg/kg/dygn fördelat på 2 till 4 doser (var 12:e till 6:e timme). Den maximala dosen bör inte överskridas med tanke på att njurar och njurfunktionen ännu inte är fullt utvecklad. Äldre: Dosjusteringar krävs ej vid normal njur- och leverfunktion. Dosering vid nedsatt njurfunktion: Till vuxna patienter med en kreatininclearance < 5 ml/min ges oförändrad rekommenderad initialdos medan efterföljande doser ska halveras med bibehållen doseringsfrekvens. Dosering vid dialys eller peritonealdialys: För patienter som genomgår hemodialys och peritonealdialys är det tillräckligt att ge 0,5-2 gram som i.v. injektion var 24 timme och efter varje dialys. Detta är effektivt nog för att behandla de flesta infektioner. Övriga rekommendationer: Observera att cefotaxim inte är effektivt vid infektioner orsakade av enterokocker. Bakteriell meningit: Vuxna rekommenderas 6- 12 g dagligen. Barn rekommenderas 150- 200 mg/kg fördelat på jämnstora doser var 6- 8:e timme. Nyfödda: Spädbarn (0- 7 dagar) ges 50 mg/kg var 12:e timme. Barn (7- 28 dagar) ges 50 mg/kg var 8:e timme. Intraabdominella infektioner: Intraabdominella infektioner bör behandlas med cefotaxim i kombination med andra lämpliga antibiotika. Sepsis: Kombination med annat passande antibiotikum bör övervägas om orsaken är gramnegativa mikroorganismer. Behandlingslängd: Cefotaximbehandlingens längd beror av patientens kliniska status och sjukdomsorsak. Cefotaxim bör administreras tills symtomen har avtagit eller tills att bevis finns för att bakterierna har eliminerats. Administreringsmetod: I.m. administrering rekommenderas enbart när i.v. infusion eller i.v. injektion inte är möjlig. Vid svåra infektioner bör Cefotaxim ges i kombination med andra antibiotika. Infusionen ska ges direkt efter beredning: - - - Intravenös infusion: För kort intravenös infusion: Efter beredning ges lösningen som en 20 minuters intravenös infusion. För lång intravenös infusion: Efter beredning ges lösningen som en 50- 60 minuter lång intravenös infusion. Upprepad intravenös injektion:. Efter beredning måste lösningen injiceras under en period på 3-5 minuter. Efter marknadsföringsgodkännandet har potentiellt livshotande arytmier rapporterats hos ett fåtal patienter som fått snabb intravenös administrering av cefotaxim genom en central venkateter. Intramuskulär injektion: Lösningen bör ges som en djup intramuskulär injektion. Lösningar som innehåller lidocain får inte ges intravenöst. Intramuskulär injektion rekommenderas inte vid svåra infektioner. Följande tabell visar lösningsvolymen för respektive injektions-/infusionsflaska: Administreringsmetod Injektionsflaskans Kort intravenös Lång intravenös Intravenös innehåll infusion infusion injektion 0,5 g 2 ml 1g 40-50 ml 4 ml 2g 40-50 ml 100 ml 10 ml Intramuskulär injektion 2 ml 4 ml - Cefotaxim och aminoglykosider bör inte blandas i samma spruta eller perfusionsvätska. 4.3 Kontraindikationer Överkänslighet mot Cefotaxim eller mot andra cefalosporinantibiotika. Tidigare omedelbar och/eller allvarlig överkänslighetsreaktion mot penicillin eller andra betalaktamantibiotika. Allergiska korsreaktioner kan förekomma mellan penicilliner och cefalosporiner (se avsnitt 4.4) 4.4 Varningar och försiktighet Som andra antibiotika kan speciellt långvarigt bruk resultera i överväxt av icke-känsliga organismer. Upprepad utvärdering av patientens tillstånd är väsentlig. Om en superinfektion uppträder under behandlingen bör lämpliga åtgärder vidtas och om det anses kliniskt nödvändigt inleda specifik antimikrobiell behandling. Anafylaktiska reaktioner Allvarliga även fatala överkänslighetsreaktioner har rapporterats hos patienter som fått cefotaxim (se avsnitt 4.3 och 4.8). Om en överkänslighetsreaktion uppträder måste behandlingen avslutas. Vid fall av allvarliga överkänslighetsrekationer eller anafylaktiska reaktioner ska den nödvändiga akutbehandlingen påbörjas enligt klinisk allvarlighetsgrad. Cefotaxin är kontraindicerad hos patienter med en tidigare historik av omedelbar överkänslighet mot cefalosporiner. Eftersom korsallergi förekommer mellan penicilliner och cefalosporiner bör användning av cefalosporiner ske med extrem försiktighet på penicillinkänsliga individer. Inledande förfrågningar angående överkänslighet mot penicillin och andra laktamantibiotika är nödvändiga före ordinering av cefalosporiner eftersom korsallergi inträffar i 5-10% av fallen. Patienter med njurinsufficiens Dosen bör justeras efter beräknad kreatininclearance (se 4.2) Försiktighet bör iaktas om cefotaxim ges tillsammans med aminoglykosider, probenecid eller andra njurtoxiska läkemedel (se avsnitt 4.5). Njurfunktionen måste övervakas hos dessa patienter, äldre och hos de med en tidigare känd njurförsämring. Allergisk diates eller astma Cefotaxim bör användas med försiktighet till patienter med allergisk diates eller astma Behandling av pneumoni Vid behandling av pneumoni ska det beaktas att cefotaxim inte är effektivt mot bakterier som orsakar atypisk pneumoni eller mot flertalet andra bakteriearter som kan orsaka pneumoni inklusive P. aeruginosa (se avsnitt 5.1). Allvarliga bullösa reaktioner Fall med allvarliga bullösa hudreaktioner som Stevens-Johnson syndrom eller toxiskt epidermal nekrolys har rapporterats med cefotaxim (se avsnitt 4.8). Patienter bör rådas att omedelbart kontakta sin läkare innan de fortsätter behandlingen om hud och/eller slemhinnereaktioner uppträder. Clostridium difficile associerad sjukdom (t.ex pseudomembranös kolit) Svår och ihållande diarré har förekommit under eller i veckorna efter behandling med Cefotaxim. Detta kan vara ett symtom på Clostridium difficileassocierad sjukdom (CDAD). CDAD kan förekomma i från mild till livshotande form. Den allvarligaste formen är pseudomembranös kolit. Diagnosen är mycket sällsynt men potentiellt fatal och tillståndet kan bekräftas genom endoskopi och/eller histologi. Det är viktigt att överväga denna diagnos hos patienter med diarré under eller efter cefotaximbehandling. Om diagnosen pseudomembranös kolit misstänks ska behandlingen med cefotaxim omedelbart avbrytas och annan lämplig behandling ska sättas in omedelbart (t.ex. speciell antibiotika/kemoterapi med styrkt klinisk effekt). Clostridium difficile associerad sjukdom kan gynnas vid fekalstas. Antiperistaltika är kontraindicerat. Hematologiska reaktioner Leukopeni, neutropeni och mera sällan agranulocytos kan utvecklas under behandling med cefotaxim, blodvärdet ska därför kontrolleras om behandlingen varar längre än 7 dagar. Behandlingen ska avbrytas om neutropeni uppkommer (< 1400 neutrofila leukocyter/mm3). Några fall av eosinofili och trombocytopeni har rapporterats vara snabbt reversibla vid behandlingsstopp. Fall med hemolytisk anemi har också rapporterats (se avsnitt 4.8). Nervtoxicitet Höga doser med betalaktamantibiotika, inklusive cefotaxim till patienter med njurinsufficiens kan orsaka encefalopati (dvs påverkan på medvetandet, onormala rörelser och kramper) (se avsnitt 4.8. Patienterna bör rådas att omedelbart kontakta sin läkare innan behandlingen avslutas om sådana reaktioner inträffar. Försiktighet vid administrering Aminoglykosider och cefotaxim bör inte blandas i samma spruta eller perfusionsvätska. Efter marknadsföringsgodkännandet har potentiellt livshotande arytmier rapporterats hos några enstaka patienter som fått snabb intravenös administrering av cefotaxim genom en central venkateter. Den rekommenderade injektions- infusionstiden bör följas (se avsnitt 4.2). Intag av natrium Natriuminnehållet i Cefotaxim (2,2 mmol/g), ska beaktas vid behandling av patienter som ordinerats begränsat natriumintag. Natriuminnehåll: 25,3 mg/50,6 mg/101,2 mg per injektionsflaska. Påverkan på diagnostiska laboratorietester Som med andra cefalosporiner har ett positivt Coombs test setts hos några patienter som behandlats med cefotaxim. Detta fenomen kan interferera vid korstestning av blod. Ett falskt positivt glukostest kan förekomma vid användning av reducerande substanser (Benedicts och Fehlings lösningar eller med Clinitest tabletter) men inte vid användning av specifika enzymbaserade tester (glukosoxidasmetoder). 4.5 Interaktioner med andra läkemedel och övriga interaktioner Med andra läkemedel: - Medel som ökar utsöndringen av urinsyra: Probenecid interagerar med den renala utsöndringen av cefotaxim, vilket ökar exponeringen av cefotaxim tvåfaldigt och minskar renalt clearance till cirka hälften vid terapeutiska doser. På grund av stort terapeutiskt index för cefotaxim krävs ingen dosjustering hos patienter med normal njurfunktion. Dosjustering kan behövas hos patienter med nedsatt njurfunktion (se avsnitt 4.4 och 4.2). - Aminoglykosidantibiotika och diuretika: I likhet med andra cefalosporiner kan cefotaxim förstärka den nefrotoxiska effekten hos nefrotoxiska läkemedel såsom aminoglykosider eller potenta diuretika (t.ex. furosemid). Njurfunktion ska övervakas hos dessa patienter (se avsnitt 4.4). - Bakteriostatiska antibiotika: Cefotaxim bör inte kombineras med bakteriostatiska antibiotika (t.ex. tetracykliner, erytromycin och kloramfenikol) eftersom en antagonistisk effekt är möjlig. Andra typer av interaktioner: - 4.6 Positivt Coombs test har setts hos patienter som behandlas med cefotaxim, liksom för andra cefalosporiner. Detta fenomen kan interferera vid korstestning av blod. Ett falskt positivt glukostest kan ses vid användning av reducerande substanser (Benedicts eller Fehlings lösning eller med Clinitest tabletter) men inte av specifika enzymbaserade tester (glukosoxidasmetoder). Graviditet och amning Graviditet Tillförlitliga säkerhetsdata av cefotaximbehandling av gravida kvinnor saknas. Djurstudier har inte visat några direkta eller indirekta skadliga effekter med avseende på reproduktionstoxicitet. Dock saknas lämpliga och välkontrollerade studier på gravida kvinnor. Cefotaxim passerar placentan. Den högsta koncentrationen mätt i amnion var högre än MIC för de flesta gramnegativa bakterier. Cefotaxim ska därför inte användas under graviditet såvida inte förväntade födelar överväger eventuella risker. Amning Cefotaxim utsöndras i bröstmjölk. Effekter på den fysiologiska tarmfloran hos det diande barnet som kan leda till diarré, kolonisering av jästliknande svampar och sensibilisering av barnet kan inte uteslutas. Ett beslut måste därför fattas om amningen eller behandlingen ska avbrytas, med beaktande av fördelarna med amning för barnet och av hur viktig behandlingen är för kvinnan. 4.7 Effekter på förmågan att framföra fordon och använda maskiner Det finns inget stöd för att cefotaxim direkt skulle försämra förmågan att framföra fordon och använda maskiner. Cefotaxim i höga doser speciellt till patienter med njurinsufficiens kan orsaka encefalopati (d.v.s. påverkan på medvetandet, onormala rörelser och kramper) (se avsnitt 4.8). Patienter ska rådas att inte framföra fordon eller använda maskiner om sådana symtom uppträder. 4.8 Biverkningar Ca 5% av de behandlade patienterna kan förvänta sig biverkningar. Dessa biverkningar är för det mesta dosrelaterade och orsakas av läkemedlets farmakologiska effekt. Frekvens Mycket Vanliga Mindre Sällsynta Mycket Ingen känd vanliga (>1/100, vanliga (>1/10 000, sällsynta frekvens (kan (>1/10) <1/10) (>1/1 000, <1/1 000) (<1/10 inte beräknas <1/100) 000) från Organsystem tillgängliga data) Infektioner Oral Tillväxt av och candidaokänsliga infestationer infekorganismer tion (se avsnitt 4.4) Blodet och Leukopeni Granylocyto lymfsystemet Eusinofili peni TromboAgranulocyt cytopeni os och (se avsnitt neutropeni 4.4) Immunsystemet JarischHerxheime r* reaktion Centrala och perifera nervsystemet Kramper (se avsnitt 4.4) Hemolytisk anemi (se avsnitt 4.4) Anafylaktisk chock (se avsnitt 4.4) Anafylaktiska reaktioner Angioödem Bronkospasm Encefalopati (dvs påverkan på medvetandet , onormala rörelser (se avsnitt 4.4) yrsel och trötthet (efter administrering av höga doser) Huvudvärk Hjärtat Magtarmkanalen Lever och gallväggar Hud och subkutan Arytmier (efter snabb bolusinfusion via en central venkateter) Illamåen Aptitlöshet de Kräknin g Magont Diarré Ökning av leverenzymer (ASAT, ALAT, LDH, gamma-GT och /eller alkaliskt fosfatas) och/eller bilirubin Utslag Klåda Pseudomembranös kolit (se avsnitt 4.4) Ikterus, hepatit* Erythema multiforme, vävnad Nässelutslag Njurar och urinvägar Tempor är ökning av serumkreatinin (speciell t när det förskrivs tillsammans med aminoglykosid er) och serumurea Allmänna I.m. Feber symtom administr Inflamoch/eller ation; mationer symtom från Smärta på administrering på injektio sstället injektion nssstället stället, inklusiv e flebiter/ tromboflebiter exudativum Stevens Johnsons syndrom Toxisk epidermal nekrolys (se avsnitt 4.4) Akut interstitiell nefrit * Efter marknadsföringsgodkännandet Jarisch- Herxheimer reaktion Vid behandling av borellios, kan Jarisch-Herxheimer reaktion utvecklas under de första behandlingsdagarna. Uppträdandet av en eller fler av följande symtom har rapporterats efter flera veckors borelliosbehandling: Hudutslag, klåda, feber, leukopeni, ökning av leverenzymer, svårighet att andas samt ledbesvär. Lever och gallvägar Ökning av leverenzymer (ALAT, ASAT, LDH, gamma-GT och eller alkalisk fosfatas) och/eller bilirubin har observerats. Dessa avvikande laboratoriefynd överstiger sällan två gånger den övre gränsen för normalvärdet och antyder tecken på leverskada som vanligtvis är cholestatisk och vanligtvis utan symtom Rapportering av misstänkta biverkningar Det är viktigt att rapportera misstänkta biverkningar efter att läkemedlet godkänts. Det gör det möjligt att kontinuerligt övervaka läkemedlets nytta-riskförhållande. Hälso- och sjukvårdspersonal uppmanas att rapportera varje misstänkt biverkning till Läkemedelsverket, Box 26, 751 03 Uppsala (http://www.lakemedelsverket.se). 4.9 Överdosering Symtom på förgiftning: Cefotaxim har en bred säkerhetsmarginal. Inga fall av akut intoxikation med cefotaxim har publicerats. Symtom på överdosering motsvarar ofta biverkningsprofilen. Vid överdosering (speciellt vid njurinsufficiens) kan reversibel encefalopati förekomma. Behandling av överdosering: Vid överdosering måste behandlingen med cefotaxim avslutas och understödjande behandling påbörjas vilket inkluderar åtgärder som ökar eliminationen samt symtomatisk behandling av biverkningarna (dvs kramper). Det finns ingen specifik antidot vid överdosering. Serumnivåerna av cefotaxim kan reduceras med hemodialys eller peritonealdialys. 5 FARMAKOLOGISKA EGENSKAPER 5.1 Farmakodynamiska egenskaper Famakoterapeutisk grupp: Cefalosporiner och liknande antibakteriella medel. ATC-kod: J01DD01 Verkningsmekanism: Cefotaxim är ett tredje generationens bredspektrum-cefalosporinantibiotikum med baktericid effekt. De baktericida egenskaperna hos cefotaxim uppkommer genom hämning av cellväggssyntesen. Resistensmekanism: - Effektiv hydrolysering av cefotaxim genom produktion av bredspektrumbetalaktamas. - induktion av/eller konstitutivt uttryck av AmpC-enzymer - impermeabilitet - effluxpumpsmekanismer - minskad affinitet för penicillinbindande proteiner till cefotaxim Fler än en av dessa möjliga mekanismer kan samexistera i en bakterie. Brytpunkter: Nuvarande MIC brytpunkter som används för att tolka känsligheten för cefotaxim. European Committee on Antimicrobial Susceptibility Testing (EUCAST), Kliniska MIC Brytpunkter (V1.1,31/03/2006) Känsliga (<)/ Resistenta(>) 1/2 --Obs3 -0,5/0,54 0,5/24 0,12/0,124 Enterobacteriaceae2 Pseudomonas Acinetobacter Staphylococcus3 Enterococcus Streptococcus A, B, C och G Streptococcus pneumoniae: Haemophilus influenzae Moraxella Catarrhalis Neisseria gonorrhoea 0,12/0,124 Neisseria Meningitides 0,12/0,124 Gramnegativa, anaerober -1 Icke artrelaterade brytpunkter 1/2 S</>R 1. Icke-artrelaterade brytpunkter har i huvudsak bestämts baserat på PK/PD-data och är oberoende av MIC fördelningen för specifika arter. De är användbara endast för arter som inte fått en artspecifik brytpunkt och inte för de arter där känslighetstest inte rekommenderas (markerade med --eller IE i tabellen). 2. Cefalosporins brytpunkter för Enterobacteriaceae kommer att uppvisa resistens medierad av de flesta ESBLs och andra kliniskt viktiga betalaktamaser i Enteriobacteriaceae. Emellertid kan några ESBL-producerande stammar vara känsliga eller intermediärt känsliga vid dessa brytpunkter. Laboratorier kan vilja använda en test som specifikt screenar närvaro av ESBL. 3. Känsligheten hos staphylococci för cefalosporiner är hämtad från methicillin (med undantag av ceftazidim som inte bör användas vid stafylokockinfektioner). 4. Stammar med MIC värden över S/I brytpunkten är mycket sällsynta eller ännu inte rapporterade. Identifieringen och antimikrobiella känslighetstester på varje sådant isolat måste upprepas och om resultatet bekräftas måste isolatet sändas till ett referenslaboratorium. Innan det finns bevis angående kliniskt svar för bekräftade isolat med MIC värden över nuvarande resistensbrytpunkter (i kursiv stil) bör de rapporteras som resistenta. -- = Test rörande känslighet rekommenderas inte eftersom arten är dåligt mottaglig för behandling med läkemedlet IE = Det finns inte tillräckligt med bevis att arten ifråga är lämplig för behandling med läkemedlet. RD = Principdokument som listar data som används av EUCAST för att bestämma brytpunkter. Känslighet Resistensprevalensen för förvärvad resistens varierar geografiskt och över tid för utvalda arter. Lokal resistensinformation är därför önskvärd speciellt vid behandling av svåra infektioner. Experthjälp bör sökas när den lokala resistensprevalensen är sådan att nyttan av att använda ett medel, vid åtminstone några typer infektioner, är tveksam. Normalt känsliga arter Grampositiva aerober Staphylococcus aureus (MSSA) Grampositiva anaerober Streptococcus pyogenes Gramnegativa aerober Escherichia coli% Haemophilus influenzae * Haemophilus parainfluenzae* Klebsiella pneumoniae#% Moraxella catarrhalis * Morganella morganii Neisseria meningitidis* P. mirabilis% Arter där resistens kan vara ett problem Grampositiva aerober Staphylococcus aureus Staphylococcus epidermis+ Staphylococcus haemolyticus+ Staphylococcus hominis+ Gramnegativa aerober Citrobacter spp* Enterobacter aerogenes Enterobacter cloacae Proteus vulgaris Serratia marcescens Anaerober Bacteroidis fragilis Naturligt resistenta arter Grampositiva aerober Enterococcus spp. Listeria spp. Gramnegativa aerober Acinetobacter spp. Pseudomonas aeruginosa Stenotrophomonas maltophilia Anaerober Clostridium difficile Övriga: Chlamydiae spp Chlamydophila spp. Mycoplasma spp. Legionella pneumophilia * Klinisk effekt har visats för känsliga isolat för godkända kliniska indikationer. Meticillin- (oxacillin-) resistenta stafylokocker (MRSA) är resistenta mot alla kända betalaktamantibiotika inklusive cefotaxim. Penicillinresistenta Streptococcus pneumoniae visar varierande grad av korsresistens mot cefalosporiner som cefotaxim. + I åtminstone en region är resistensgraden över 50 % # I intensivvårdsenheter är resistensgraden >10 % % Extended Spectrum Beta-Laktamas (ESBL) producerande stammar är alltid resistenta. 5.2 Farmakokinetiska uppgifter Absorption Cefotaxim administreras parenteralt. Intravenös bolusinjektion av 1 gram cefotaxim ger en maximal koncentration på ca 81-102 mg/l efter 5 minuter, intravenös bolusinjektion av 2 g cefotaxim ger en maximal koncentration på 167-214 mg/l efter 8 minuter. Intramuskulär injektion av 1 gram cefotaxim ger en maximal plasmakoncentration på 20 mg/l inom 30 minuter. Distribution Cefotaxim penetrerar väl in i olika distributionsrum. Terapeutiska läkemedelsnivåer som överstiger vanliga patogeners minsta hämmande koncentration (MIC-värden) uppnås snabbt. Koncentrationen i cerebrospinalvätskan är låg när hjärnhinnorna inte är inflammerade, men är mellan, 3-30 µg/ml hos barn med hjärnhinneinflammation. Cefotaxim passerar normalt blodhjärnbarriären i nivåer över MIC, för normalkänsliga patogener, vid hjärnhinneinflammation. Cefotaximkoncentrationer (0,2-5,4 µg/ml) som hämmar de flesta gramnegativa bakterier uppnås i varigt sputum, bronkialsekret och pleuravätska efter doser på 1 och 2 gram. Koncentrationer som troligen är effektiva mot känsliga organismer uppnås efter terapeutiska doser i interstitiell vätska, njurvävnad, peritonealvätska och i gallblåsans vägg. Höga koncentrationer av cefotaxim och O-desacetylcefotaxim återfinns i gallan. Cefotaxim passerar placentan och uppnår höga koncentrationer i fostervätska och fostervävnad (upp till 6 mg/kg). Små mängder cefotaxim utsöndras i modersmjölken. Proteinbindningsgraden för cefotaxim är ca 25-40 %. Den skenbara distributionsvolymen för cefotaxim är 21-37 liter efter en 30 minuters intravenös infusion av 1 gram cefotaxim. Biotransformation: Cefotaxim metaboliseras delvis hos människa. Ca 15-25% av en parenteral dos metaboliseras till O-desacetylcefotaxim som också är mikrobiologiskt aktivt. Elimination: Cefotaxim och O-desacetylcefotaxim utsöndras huvudsakligen via njurarna. Endast en liten del cefotaxim (2%) utsöndras i gallan. 40-60% av administrerad cefotaximdos återfinns i oförändrad form i urinen inom 6 timmar och 20% återfinns som O-desacetylcefotaxim. Efter administrering av radioaktivt märkt cefotaxim återfinns mer än 80% i urinen, 50-60% av detta som oförändrad cefotaxim och resten som metaboliter. Cefotaxims totala clearance är 240-390 ml/min och dess renala clearance 130-150 ml/min. Halveringstiden i serum är normalt ca 50-80 minuter för cefotaxim och 90 minuter för Odesasacetylcefotaxim. Hos äldre patienter är halveringstiden i serum 120-150 min. Hos patienter med nedsatt njurfunktion (kreatininclearance 3-10 ml/min) kan halveringstiden för cefotaxim i serum öka till 2,5-3,6 timmar. Hos nyfödda påverkas farmakokinetiken av graviditetens längd samt kronologisk ålder. Halveringstiden är förlängd hos förtidigt födda och nyfödda med låg födelsevikt. 5.3 Prekliniska säkerhetsuppgifter Gängse studier avseende allmäntoxicitet, gentoxicitet, och reproduktionseffekter visade inte några särskilda risker för människa. 6 FARMACEUTISKA UPPGIFTER 6.1 Förteckning över hjälpämnen Inga 6.2 Inkompatibiliteter Cefotaxim ska inte blandas med andra antibiotika i samma spruta eller infusionslösning, speciellt inte med aminoglykosider. Cefotaxim Stragen ska inte blandas med lösningar innehållande natriumbikarbonat. 6.3 Hållbarhet Pulver till injektionsvätska, lösning: 3 år. Pulver till injektions- och infusionsvätska, lösning: 3 år. Efter första öppnandet/rekonstitution: Kemisk och fysikalisk stabilitet vid användning har visats under 12 timmar vid 25 ºC och under 24 timmar vid 2-8 ºC. Ur mikrobiologisk synvinkel ska produkten användas omedelbart. Om den inte används omedelbart är användningstiden och lagringsförhållandena användarens ansvar och normalt bör denna tid inte vara längre än 24 timmar vid 2-8 ºC, såvida rekonstitutionen/utspädningen inte har utförts under kontrollerade och validerade förhållanden. 6.4 Särskilda förvaringsanvisningar Inga särskilda förvaringsanvisningar. Förvaras i originalförpackningen. Ljuskänsligt. För förvaringsanvisningar för rekonstituerad/utspädd produkt, se avsnitt 6.3. 6.5 Förpackningstyp och innehåll Förpackningstyp 0,5 g pulver till injektionsvätska, lösning: 10 ml injektionsflaska av klart glas, Ph. Eur. typ III, med halogenerad butylgummipropp typ I. 1 g pulver till injektions- och infusionsvätska, lösning: 10 ml injektionsflaska av klart glas, Ph. Eur. typ III, med halogenerad butylgummipropp typ I. 2 g pulver till injektions- och infusionsvätska, lösning: 20 ml injektionsflaska av klart glas, Ph. Eur typ I, med halogenerad butylgummipropp typ I. eller 50 ml injektionsflaska av klart glas, Ph. Eur. typ I, med halogenerad butylgummipropp typ I Förpackningsstorlek 10 injektionsflaskor per kartong Eventuellt kommer inte alla förpackningsstorlekar att marknadsföras. 6.6 Anvisningar för användning och hantering samt destruktion För att undvika risk för infektioner ska beredningen av lösningen ske med aseptisk teknik. Vid kort intravenös infusion kan 1 g eller 2 g Cefotaxim Stragen lösas i 40-50 ml av en av de kompatibla lösningsmedlen. Vid lång intravenös infusion kan 2 g Cefotaxim Stragen lösas i 100 ml av en av de kompatibla lösningsmedlen. Vid intravenös injektion bör Cefotaxim Stragen 0,5 g lösas i 2 ml vatten för injektionsvätskor, Cefotaxim Stragen 1 g bör lösas i 4 ml vatten för injektionsvätskor och Cefotaxim Stragen 2 g bör lösas i 10 ml vatten för injektionsvätskor. Vid intramuskulär injektion bör Cefotaxim Stragen 0,5 g lösas i 2 ml vatten för injektionsvätskor eller Cefotaxim Stragen 1,0 g löses i 4 ml vatten för injektionsvätskor. För att förhindra smärta orsakad av injektionen kan Cefotaxim Stragen 0,5 g lösas i 2 ml 1% lidokainlösning eller Cefotaxim Stragen 1,0 g kan lösas i 4 ml 1% lidokainlösning (enbart till vuxna). Kompatibla lösningsmedel för rekonstitution: - Vatten för injektionsvätskor - Natriumklorid 9 mg/ml (0,9%) - Glukos 50 mg/ml (5%) - Lidokain 1% (endast för intramuskulärt bruk) Efter beredning ska lösningen vara klar och ljusgul till gulbrun. Använd inte lösningen om det finns synliga partiklar i lösningen. Endast för engångsbruk. Ej använt läkemedel och avfall ska kasseras enligt gällande anvisningar. Se 4.2 för beredningsinstruktioner. 7 INNEHAVARE AV GODKÄNNANDE FÖR FÖRSÄLJNING Stragen Nordic A/S Helsingørsgade 8C DK- 3400 Hillerød Danmark 8 NUMMER PÅ GODKÄNNANDE FÖR FÖRSÄLJNING Pulver till injektionsvätska, lösning: 21297 Pulver till injektionsvätska och infusionsvätska, lösning: 21298 9 DATUM FÖR FÖRSTA GODKÄNNANDE/FÖRNYAT GODKÄNNANDE 2004-12-22 / 2009-05-24 10 DATUM FÖR ÖVERSYN AV PRODUKTRESUMÉN 2016-07-14