1 Uppgifter - UU Studentportalen
advertisement

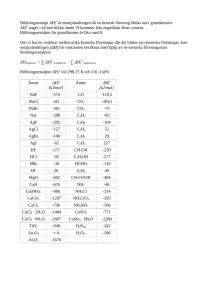
1 Uppgifter 1.1 Steady-state-approximationen − I en natriumnitratsmälta sönderfaller bromatjoner BrO− 3 i bromidjoner Br och syrgas O2 . Kinetiska undersökningar antryder att mekanismen för sönderfallet är − − k1 -→ BrO− BrO− 2 + BrO (långsam) 3 + Br k2 − BrO− 2 -→ Br + O2 (mycket snabb) k3 − -→ 2BrO− BrO− 2 (mycket snabb) 3 + BrO Härled, med lämpligt valda approximationer, uttryck för (a) förbrukningshastigheten för BrO− 3 , (b) bildningshastigheten för O2 . c. Mätning av hastigheten r för bromatjonsönderfallet vid två olika temperaturer gav följande resultat. Beräkna aktiveringsenergin för steg 1 i mekanismen ovan. 1.2 T /K [BrO− 3] / M Br− / M r / M/min 643 683 0.0443 0.0172 0.519 0.759 3.22 × 10−4 5.09 × 10−4 Fotokemi I ett fotokemiskt experiment skapar man med en kort laserblixt (ca 5 ns) exciterat singlett-tillstånd i en molekyl A. Singletten deaktiveras mycket snabbt med kvantutbytet φisc = 0.76 via “intersystem crossing” till det lägsta triplettexciterade tillståndet. Detta deaktiveras i sin tur genom fosforescens (radiativt) med hastighetskonstanten kp = 5 × 105 s−1 och ickeradiativt knr (ej känd) till grundtillståndet. Livslängden för tripletten upmättes under dessa förhållanden till 1.7µs. I närvaro av syre sker utsläckning av tripletten med hastighetskonstanten kq varvid singlettsyre 1 O2 bildas. Experimentet utförs i vattenlösning. Reaktionerna kan sammanfattas i följande schema: A∗ -→ AT (φisc = 0.76) kp AT -→ A + hν knr AT -→ A kq AT + O2 -→ A +1 O2 Lösligheten för syre i vatten vid 1 atm och 20 ◦ C är enligt http://water.usgs.gov/owq/FieldManual/Chapter6/table6.2_6.pdf 9.1 mg/l. Under dessa förhållanden är livslängden för tripletten 1300 ns. Beräkna hastighetskonstanten kq för utsläckningen av tripletten med syre samt kvantutbytet för bildningen av singlettsyre. 1 1.3 Relaxationsmetoder Den reversibla elementarreaktionen kf * CH3 COO− + H+ − ) − CH3 COOH (1) kb studerades med hjälp av temperatursprångsmätningar i avsikt att bestämma hastighetskonstanterna kf och kb vid 25.0◦ C. Starttemperaturen och temperatursprångets storlek avpassades så att sluttemperaturen blev just 25.0◦ C. För en provlösning som framställts genom att i vatten lösa upp 0.0100 mol ättiksyra i en mätkolv med volymen 1000 ml uppmättes relaxationstiden till 2.7 × 10−8 s. Vid 25.0◦ C är den stökiometriska jämviktskonstanten Kc för (1) värdet 5.5 × 104 l/mol. Beräkna hastighetskonstanterna kf och kb . Vattnets autoprotolys kan försummas. 1.4 Aktiveringsenergier För totalreaktionen 2NO + O2 -→ 2NO2 i gasfas har följande mekanism föreslagits: k1 2NO -→ N2 O k2 N2 O2 -→ 2NO k3 N2 O2 + O2 -→ 2NO2 (1) (2) (3) Visa att bildningshastigheten för NO2 följer hastighetsuttrycket 2k1 k3 [NO]2 [O2 ] d[NO2 ] = dt k2 + k3 [O2 ] Fortvarighetstillståndsapproximationen kan tillämpas på den reaktiva mellanprodukten N2 O2 . app Beräkna vidare den skenbara aktiveringsenergin Ea för totalreaktionen under förutsättning att endast en liten del av den i (1) bildade N2 O2 reagerar med O2 enligt (3). Resten återgår i (2) till NO. Aktiveringsenergierna för stegen (1)–(3) är 82, 205 respektive 82 kJ/mol. 1.5 Experimentella metoder, reaktionsordning och aktiveringsenergier För att bestämma reaktionshastigheter används ett flertal olika experimentella metoder. Inom biokemin är stopped-flow-tekniker vannligt förekommande. I ett stopped-flow-experiment för att bestämma kinetiska parametrar för DNA-inbindningen av ett malarialäkemedel uppmättes för associationen och dissociationen följande data. Vid den våglängd som användes vid 2 mätningen absorberar endast läkemedlet, dock i både bunden och fri form (f r itt = 5500 M−1 cm−1 ). Den optiska väglängden i försöket var 1 cm. Reaktionen kan skrivas k+ −* P +D ) − PD k− där P är fritt DNA, D fritt läkemedel, och P D DNA:läkemedel-komplexet. Jämviktkonstanten för reaktionen ovan är mycket hög. Association, 20 ◦ C, [DNA]tot =100µM. Vid t = 0 finns endast fritt (obundet) läkemedel i provet. t/ms A 0 100 200 400 800 1000 0.55 0.50 0.45 0.40 0.33 0.31 I ett parallellt experiment, med samma halt läkemedel, och med samma optiska väglängd, men med stort överskott på DNA, uppmättes absorbansen A = 0.15 efter lång tid. För att observera dissociationen sattes SDS till en P D-lösning vilket kvantitativt driver reaktionen åt fritt DNA och fritt läkemedel (K ≈ 0). I stoppedflow-apparaten uppmättes följande halveringstid för P D vid olika temperaturer. (För en viss temperatur var alla halveringstider lika.) Dissociation T /◦ C t1/2 / min 1 5 10 20 30 0.65 0.52 0.40 0.24 0.15 1. För jämvikter som inte är så kraftigt förskjutna åt ena hållet som reaktionen ovan kan man använda s.k. relaxationsmetoder för att studera kinetiken. Ge en kortfattad beskrivning av principen bakom temperatursprångsmätningar (“T-jump”). (2p) 2. Avgör vilken reaktionsordning associationen har, och beräkna hastighetskonstanten. (4p) 3. Beräkna aktiveringsenergin för dissociationen. (4p) 1.6 Relaxationsmetoder 2 En reaktion k+ * A− ) −B k− studerades med hjälp av relaxationstidsmätningar. Följande hastighetskonstanter och jämviktskonstanter erhölls vid olika temperaturer: 3 T /◦ C kobs /s −1 K 5 10 20 40 80 10.04 12.78 20.25 47.00 192.7 1.30 1.37 1.51 1.81 2.43 a Beskriv principen för hur man med relaxationsmetoder kan bestämma kinetiska data. (2p) b Beräkna hastighetskonstanterna k+ och k− vid de olika temperatorerna. (3p) c Beräkna aktiveringsenergier och pre-exponentiella faktorer för framoch backreaktionerna, samt skissa ett kvalitativt korrekt energidiagram för reaktionen. (3p) d I framreaktionen bryts en O − H-bindning. Kommer k+ att påverkas av att vätet byts ut mot deuterium, och i så fall hur? Förklara eventuella effekter. (2p) 1.7 Enzymkinetik Några glada biokemister uppmätte för en enzymreaktion följande data vid 300 K. Totalhalten enzym var [E]0 = 5.9µM. [S]0 /µM r /M −1 s −1 10 20 50 100 1000 5 000 0.0294541 10 000 0.029477 0.016573 0.021223 0.025519 0.0273655 0.0292717 Vidare försökte biokemisterna, vilka inte hade varit så uppmärksamma på föreläsningarn i kinetik, att bestämma aktiveringsenergin för reaktionen E + S -→ E + P genom att vid för olika [E]0 och [S]0 mäta r (vid konstanta koncentationer) som funktion av T och rita upp ln r mot 1/T . Till sin stora förvåning fann man att aktiveringsenergin berodde på [S]0 och [E]0 , men att den verkade konvergera vid höga [S]0 . a Följer reaktionen Michaelis–Menten-kinetik? Om ja, beräkna i så fall KM och kb . (5p) b Förklara varför reaktionshastigheten i Michaelis–Menten-kinetik går mot ett gränsvärde vid höga [S]0 . (1p) c Vad innebär kompetitiv inhibering? (2p) d Förklara varför den uppmätta aktiveringsenergin inte är oberoende av [S]0 . (2p) 4 2 Lösningar 2.1 Steady-state-approximationen − d[Br O3− ] = k1 [Br O3− ][Br − ] + k3 [Br O3− ][Br O − ] dt Gör steady-state-antagande på [BrO− ]: d[Br O − ] = k1 [Br O3− ][Br − ] − k3 [Br O3− ][Br O − ] = 0 dt [Br O − ] = − k1 [Br O3− ][Br − ] k1 = [Br − ] k3 [Br O3− ] k3 d[Br O3− ] k1 = k1 [Br O3− ][Br − ] + k3 [Br O3− ] [Br − ] = 2k1 [Br O3− ][Br − ] dt k3 Bildningshastighet för syrgas: d[O2 ] = k2 [Br O2− ] dt Gör steady-state-antagande på [BrO− 2 ]: d[Br O2− ] = k1 [Br O3− ][Br − ] − k2 [Br O2− ] + 2k3 [Br O3− ][Br O − ] = 0 dt Br O2− = 3k1 [Br O3− ][Br − ] k2 k2 3k1 [Br O3− ][Br − ] d[O2 ] = k2 [Br O2− ] = = 3k1 [Br O3− ][Br − ] dt k2 − Alternativ lösning: Eftersom BrO− 3 sönderfaller i Br och 3/2O2 måste syrgasen bildas 3/2× så fort som bromatjonerna förbrukas. Aktiveringsenergin fås ur Ea = R ln 1 k1 (T1 ) k1 (T2 ) 1/T2 − 1/T1 Hastighetskonstanten vid de två temperaturerna ges av k1 = vilket insatt i Arrheniusuttrycket ger Ea = 93.5 kJ/mol. 2.2 r , 2[Br O3− [Br − ] Fotokemi Under vanliga fotokemiska reaktionsförhållanden är halten av exciterat tillstånd mycket låg. Detta gör att syre ([O2 ] = 0.28 mM) är i stort överskott, och utsläckningsreaktionen kan behandlas som en pseudo-1:a-ordningens reaktion. Ur livslängdsdata får vi att summan av alla deaktiverande hastighetskonstanter i frånvaro av syre är (1.7 × 10−6 )−1 = 588 000 s−1 . Med syre 5 i lösningen ökar deaktiveringshastigheten till 769 000 s−1 . Syreutsläckningens bidrag är skillnaden i effektiv deaktiveringshastighetskonstant, d.v.s. kq [O2 ] = 181 000 s−1 , vilket med den givna syrehalten ger kq = 6.4 × 108 M−1 s−1 . Kvantutbytet för singlettsyrebildningen är reaktionshastigheten för singlettsyrereaktionen genom den totala deaktiveringshastigheten. Dessutom måste vi ta hänsyn till att endast 76% av alla absorberade fotoner leder till bildning av triplett. Kvantutbytet blir alltså φq = kq [O2 ]τO2 × 0.76 = 0.18, där τO2 är livslängden i närvaro av syre. Notera att kvantutbytet är koncentrationsberoende. 2.3 Relaxationsmetoder Låt acetat=A, protoner=B och ättiksyra=C. Vid jämvikt är fram- och backreaktionerna lika snabba: 0 = r = kf [A]eq [B]eq − kb [C]eq vilket ger att den stökiometriska jämviktskonstanten Kc = kf /kb . Totalhalten C är 0.0100 mol. För varje mol som protolyseras bildas 1 mol A och 1 mol B. Om vattnets autoprotolys försummas får vi [A]eq = [B]eq och [C]eq = [C]0 − [A]eq . Tillsammans med jämviktsuttrycket ger detta 0.01 − [A]eq [A]2eq = 5.50 × 104 ur vilket man får [A]eq = 4.17 × 10−4 M. Relaxationstiden för jämviktsinställningen följer sambandet τ −1 = kf (Aeq + Beq ) + kb . Med jämviktskoncentrationerna och jämviktskonstanten uttryckt i kf och kb får man kf = kb ×Kc och (2.7×10−8 )−1 = (5.50×104 ×2×4.17× 10−4 + 1)kb . Detta ger kb = 7.9 × 105 s−1 och kf = 4.3 × 1010 M−1 s−1 . Notera att protoneringen är utomordentligt snabb. Detta är på grund av protonens mycket höga rörlighet i vattenlösningar. 2.4 Aktiveringsenergier Vid fortvarighetstillstånd gäller att 0= d[N2 O2 ] = k1 [NO]2 − k2 [N2 O2 ] − k3 [N2 O2 ][O2 ] dt och följaktligen är [N2 O2 ]ss = k1 [NO]2 k2 + k3 [O2 ] Bildningshastigheten för produkten NO2 i steg tre är d[NO2 ] = 2k3 [N2 O2 ]ss [O2 ] dt Notera tvåan framför k3 ! Med uttrycket för fortvarighetshalten av N2 O2 ger detta d[NO2 ] 2k1 k3 [NO]2 [O2 ] = dt k2 + k3 [O2 ] 6 v.s.b. Enligt uppgiften reagerar endast en liten del av den bildade N2 O2 via (3). Detta innebär att k2 [N2 O2 ] k3 [N2 ][O2 O2 ]. Med denna förenkling blir uttrycket för bildningshastigheten 2k1 k3 [NO]2 [O2 ] d[NO2 ] = dt k2 Vidare vet vi att hastighetskonstanter kan tecknas som k = A exp(−Ea /RT ). Den skenbara aktiveringsenergin för reaktionen ovan ges av den skenbara hastighetskonstantens temperaturberoende: d ln k app = −Ea /R d(1/T ) Logaritmering av den sammansatta hastighetskonstanten ovan och derivering med avseende på 1/T ger vid handen att den skenbara aktiveringsenergin är app Ea = Ea,1 + Ea,3 − Ea,2 = −41kJ/mol Observera att en skenbar aktiveringsenergi mindre än noll inte är något konstigt för en komplex reaktion. För uppgiftens mekanism betyder det att jämvikten i reaktion (1) och (2) förskjuts mer åt vänster än reaktionshastigheten för (3) ökar. 2.5 Experimentella metoder, reaktionsordning och aktiveringsenergier 1. Vi vet omedelbart inte reaktionsordningen eller reaktanternas halter. Följande information ges dock i uppgiften (kalla läkemedlet D, DNA för P ). Index f betecknar fritt, b bundet, och 0 totalhalt. (a) Vid tiden t = 0 är halten [D]f = [D]0 och At=0 = 0.55. Vidare är f = 5500 cm−1 M−1 och l = 1 cm. Detta ger att [D]t=0 = [D]0 = 0.55/5500 = 100 × 10−6 M. (b) Vid lång tid (reaktionen har gått till jämvikt) och stort överskott DNA (allt D föreligger som D · P ) är absorbansen, med samma halter och väglängder, lika med 0.15. Detta ger att b = 1500 cm− 1M− 1. (c) Vid varje given tidpunkt är den totala absorbansen summan av bidragen från fritt och bundet D. A = f [D]f l + f [D]f l. Halten fritt D kan då skrivas som [D]f = A − b l[D]0 f l − b l (d) Eftersom reaktanthalterna är ungefär lika stora bör [D] uppvisa en koncentrationsprofil motsvarande reaktionsordningen. (e) Antag att reaktionen är av 2:a ordningen totalt. Då skall 1/[D]f mot t ge en rät linje med lutningen k+ . (k− är så litet att backreaktionen kan försummas.) Grafen blir mycket riktigt rät, och har lutningen 15044 ≈ 15000 M−1 cm−1 . 7 2. Om alla halveringstider är lika (vid en fix temperatur) är reaktionen av 1:a ordningen. Hastighetskonstanten ges då av k− = ln 2/t1/2 . En graf av ln 2/t1/2 mot 1/T ger en rät linje med lutningen −4210 K, vilket ger Ea = 35 kJ/mol. 2.6 Relaxationsmetoder 2 kobs = k+ + k− och K = k+ /k− . Detta ger att k+ = Kk− , och följakligen k− = kobs /(1 + K), och k+ = kobs − k− . T /c ir cC k1 /s −1 k2 /s −1 5 10 20 40 80 5.67403 7.39192 12.2103 30.261 136.523 4.36184 5.39462 8.07407 16.7411 56.1709 En graf av ln k mot 1/T ger lutningar på −4162 resp. −3343, vilket ger Ea1 = 34600 J/mol och Ea2 = 27800 J/mol. A1 = 1.8 × 107 s−1 , A2 = 7.4 × 105 s−1 . 2.7 Enzymkinetik Med uppgiftens siffror ger en graf av 1/r mot 1/[S]0 en rät linje (se figur) med lutningen 0.0002644 = KM /rmax . Avskärningen med x-axeln är i −1.28 × 105 = −1/KM , med y-axeln i 33.9 = 1/rmax . Följaktligen är rmax = 0.0295003 = kb [E]0 , vilket ger kb = 5000s −1 , och KM = 0.0002644∗rmax = 7.8 × 10−6 M. En graf av ln r mot 1/T innehåller vid låga [S]0 aktiveringsenergierna för alla reaktionerna (vilket bl. a. ger temperaturberoendet för KM ). Vid höga kb [S]0 är det hastighetsbegränsande steget ES -→ P + E, och man kan extrahera den egentliga aktiveringsenergin för produktbildningen. 8 60 50 1/r/ (s/M) 40 30 Function: line Coefficient values a =33.898 b =0.00026441 20 10 0 3 -100x10 -50 0 1/[S]0 / (1/M) 9 50 100