Skrivmall 11-03
advertisement

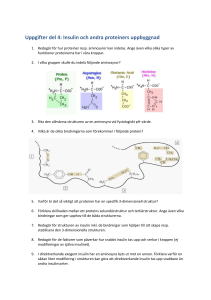
2012-02-06 3 Reglering av insulinsekretionen Åke Sjöholm Inledning Typ 2-diabetes (T2D), med en prevalens på 5–10 procent i större delen av västvärlden, är ett betydande socioekonomiskt hälsoproblem som bara ökar i takt med en åldrande och allt fetare befolkning. Sjukdomen karakteriseras av en absolut eller relativ brist på insulin, hormonet med de starkaste hypoglykemiska egenskaperna. Trots massiva forskningsinsatser sedan insulinets upptäckt 1922 har man ännu inte kunnat påvisa någon klar etiologisk faktor för diabetes. För att få en mer fullständig bild av sjukdomens orsaker är det nödvändigt att först kartlägga vilka mekanismer som kontrollerar insulinfrisättningen, såväl den akuta insulinsekretionen och insulinets biosyntes som långsiktiga förändringar i insulinproduktionen, från den friska betacellen. Denna cell kan uppfattas som en unik ”mikrosensor” av nivån glukos i blod, och bevarandet av normal glukostolerans förutsätter att hormonfrisättningen från denna cell fungerar optimalt. Skulle betacellen svikta i detta avseende, uppkommer glukosintolerans och så småningom kliniskt manifest diabetes. Insulinfrisättningens anatomi De insulinproducerande betacellerna utgör en dominerande celltyp i de langerhanska öarna, små mikroorgan som ligger utspridda i pankreas. 2012-02-06 Ungefär 1–3 miljoner öar, med en diameter vanligen mellan 50 och 1 000 µm, finns i en normal human pankreas. Jämfört med det exokrina körtelparenkymet utgör den endokrina delen (de langerhanska öarna) bara 1–2 procent. I flertalet djurarter utgör under friska förhållanden betacellerna öarnas själva kärna (60–80 procent av öarna) och omges av en mantel av andra endokrina celltyper. Huruvida detta är fallet även hos människa är ännu omtvistat. Vid utveckling av T2D sker en gradvis förlust av betaceller och man brukar räkna med att cirka hälften av betacellerna gått förlorade vid den tidpunkt T2D diagnostiseras (1). Detta utgör en del av det progressiva naturalförloppet av T2D. Förutom insulin, produceras i öarna även andra hormoner med viktiga roller i glukosmetabolismen: glukagon som produceras i alfacellerna är insulinets motsats, ett katabolt hormon som höjer blodsockernivån framför allt genom att mobilisera glukos från levern och muskulatur; somatostatin som bildas i D-cellerna är en generell hämmare av hormoners (bland annat insulin) sekretion; amylin som tillsammans med insulin (men i avsevärt mindre mängder) produceras i betacellerna är en 37 aminosyror lång peptid som anses hämma insulinsekretionen i en autokrin feedback-loop; ghrelin som bildas i ε-cellerna är ett aptit- och mättnadsreglerande hormon; och pankreaspolypeptid producerad i PP-cellerna som anses reglera bland annat pankreas exokrina funktion. Det sker även inom de langerhanska öarna en parakrin reglering av hormonsekretionen såtillvida att insulin (och högt glukos) hämmar frisättningen av glukagon, medan däremot glukagon ökar insulinsekretionen. Somatostatin hämmar sekretionen av både insulin och glukagon. Insulinets biosyntes: från gen till färdigt hormon Hos människa finns endast en insulingen, till skillnad från gnagare som har två. Att insulingenen transkriberas till mRNA och sedan vidare endast i 2012-02-06 betacellen beror på ett antal transkriptionsfaktorer som, via interaktion med insulingenens promotorregion, tillåter detta; i kroppens alla andra celler är uttrycket av insulin avstängt genom olika repressiva mekanismer (2). I terapeutiska sammanhang har man dock övervägt möjligheten att genom ”genetisk ingenjörskonst” låta andra celler – som saknar betacellernas autoantigen – börja producera insulin, till exempel vid typ 1-diabetes. Tanken härmed är att säkerställa insulinproduktion från celler som inte destrueras av immunsystemet. Detta har visats kunna genomföras i till exempel leverceller under vissa experimentella betingelser, men det är oklart hur välkontrollerad insulinfrisättningen är då dessa celler normalt saknar de mekanismer för glukosavkänning som betacellen är utrustad med. Insulinbiosyntesen och dess olika steg kan, liksom den glukosstimulerade insulinfrisättningen, vara en potentiell defekt vid T2DM då en ökad insulinsyntes är nödvändig för att upprätthålla hypersekretion av insulin vid olika tillstånd av ökat perifert insulinbehov (2). Insulingenens transkriptionsprodukt kallas preproinsulin-mRNA. Genom en så kallad signalpeptid binds molekylen till cellens endoplasmatiska nätverk, där ribosomal proteinsyntes av preproinsulin (bestående av 110 aminosyror) och ytterligare modifieringar sker: Från preproinsulin klipps signalpeptiden av, varvid proinsulin bildas som sedermera translokeras ut ur nätverket. Om belastningen på nätverket blir alltför hög, eller defekt protein bildas, kan betacellen reglera nedbrytning av dessa proteiner genom så kallad ”unfolded protein response” (3). I det korta perspektivet (< 3 timmar) stimulerar högt glukos insulinbiosyntesen translationellt utan de novo-syntes av preproinsulin-mRNA, medan däremot längre exponeringar även leder till förhöjningar i preproinsulin-mRNA. Noterbart är att nivåerna av preproinsulin-mRNA är minskade i T2Ddjurmodeller (2), och det anses att glukotoxicitet och lipotoxicitet bidrar till en försämrad betacellsfunktion delvis genom att minska insulingenens expression (2, 3). Förutom transkription av insulingenen, regleras även nivån av preproinsulin-mRNA av degradation och det anses att båda dessa 2012-02-06 mekanismer är lika viktiga för att upprätthålla adekvata nivåer av preproinsulin-mRNA i betacellen. Genom vesikulär transport från det endoplasmatiska nätverket kommer proinsulinet till Golgiapparaten där det förpackas i så kallade sekretgranulae, små blåsformiga korn som bildas av Golgiapparatens membran och knoppas av från densamma. I sekretgranulae kommer proinsulinet, genom enzymatisk spjälkning, att processas till moget och biologiskt aktivt insulin genom att den så kallade C-peptiden klipps av. På grund av surt pH i sekretgranulae kommer insulinet att förpackas i kristallin (olöslig) form. Kristallen består av sex insulinmolekyler sammanhållna av två zinkatomer. Vid stimulering av betacellen, med till exempel glukos, kommer transporten av sekretgranulae att accelereras. De smälter samman med cellens plasmamembran i en process kallad exocytos, varvid pH i granulae höjs och insulinet övergår i löslig form så att det kan transporteras i blodbanan. Samtidigt med insulin kommer även C-peptid (som också lagras i sekretgranulae) att släppas ut i blodet från betacellerna och används kliniskt för att uppskatta graden av endogen insulinproduktion, då C-peptid (till skillnad från insulin) inte metaboliseras i levern. På likartat sätt används ibland kvoten proinsulin/insulin kliniskt som surrogatmått på betacellsfunktion. Vid sviktande betacellsfunktion, till exempel vid T2D, frisätts oproportionerligt mycket proinsulin från betacellerna (4). Pankreas dräneras i vena porta som uppvisar höga och oscillerande (se nedan) insulinnivåer, och man räknar med att cirka 50 procent av insulinet extraheras av levern under hormonets förstahandspassage genom detta organ. C-peptid, som initialt ansågs vara en inert slaggprodukt, har föreslagits ha positiva effekter vid bland annat mikroangiopatiska diabeteskomplikationer (5). 2012-02-06 Metabolism av glukos och andra näringsämnen i betacellen stimulerar dess insulinfrisättning Mekanismerna som reglerar insulinfrisättningen från betacellen är föremål för kontroll av ett flertal inkommande signaler: näringsämnen och hormoner i blodet, nervimpulser från omgivande ganglia samt parakrina influenser från andra celltyper i de langerhanska öarna (6). De intracellulära mekanismer som medierar dessa signaler i betacellen har studerats intensivt och bland annat befunnits omfatta ökningar i den cytoplasmatiska Ca2+koncentrationen, hydrolys av fosfoinositider i plasmamembranet och bildning av cykliskt AMP. Av kroppens alla celler är betacellen sannolikt unik i så måtto att dess huvudsakliga fysiologiska stimulus, glukos, måste metaboliseras för att betacellens insulinfrisättande maskineri ska fungera. Betacellen känner av den omgivande nivån av glukos genom intracellulära metaboliter av glukos, genererade i glykolysen och Krebscykeln. Den kvantitativt viktigaste metabola vägen för glukos omfattar dess initiala upptag i betacellen från blodet genom GLUT2-transportörer belägna i plasmamembranet, följt av dess fosforylering av hexokinas och glukokinas (av somliga uppfattad som betacellens ”glukossensor”). Detta enzym har terapeutiskt intresse då det i flera läkemedelsföretags pipeline eller produktportfölj finns farmakologiska aktivatorer av glukokinas (7). Dessa har visat sig kunna motverka diabetes i djurmodeller, bland annat genom att aktivera betacellens glukokinas och därigenom öka insulinsekretionen (7). Glukokinas är även säte för mutationer i subtyper av MODY (7). Glukos genomgår ytterligare konversion till pyruvat och acetyl-CoA i glykolysen samt vidare oxidativ metabolisering i mitokondriernas Krebscykel. Sockrets oxidativa katabolism förefaller bilda signaler av essentiell betydelse för insulinfrisättningen, eftersom denna blockeras av mitokondriella inhibitorer. Glukos inducerar en akut aktivering av pyruvatdehydrogenas, ett hastighetsbegränsande steg i glukosoxidationen, liksom en långsiktig ökning i betacellens mRNA för detta enzym. Det ATP som bildas genom nedbrytningen av glukos stänger K+-kanaler i betacellens plasmamembran, 2012-02-06 vilket orsakar depolarisering med åtföljande inflöde av Ca2+ via spänningsberoende Ca2+-kanaler. Eftersom det råder en 10 000 gångers gradient mellan extracellulär och intracellulär Ca2+-koncentration, kommer det att ske ett snabbt inflöde av Ca2+ över betacellens plasmamembran. Härigenom stimuleras den pool av sekretgranulae som ligger nära eller redan dockade till plasmamembranet till snabb exocytos. Det anses att denna mekanism (den K ATP -beroende) ligger bakom den första fasens snabba och transienta insulinsekretion, vilken har till uppgift att bland annat hämma leverns glukosproduktion. Glukos förmår dock åtminstone delvis stimulera insulinfrisättningen oberoende av denna mekanism (8) och det anses att denna K ATP -oberoende insulinsekretion utgör grunden för den andra fasens långsamma och långdragna insulinsekretion som åstadkoms genom mobilisering av den reservpool av sekretgranulae som ligger långt ifrån plasmamembranet. De sekretionsbefrämjande signalerna i glykolysen kan antas stimulera vidare mitokondriell oxidation eller anapleros, men deras exakta natur har förblivit okänd. Det verkar dessutom som om två metabola signaler, från proximala glykolysen samt mitokondrien, sammanfaller för att potentiera insulinsekretionen. Vid human T2D är betacellens insulinproduktion defekt, och det är framför allt den glukosstimulerade insulinsekretionen som är nedsatt (9). Endokrina pankreas blodflöde reglerar insulinsekretionen De langerhanska öarna är rikligt genomblödda och svarar för cirka 10 procent av hela pankreas blodcirkulation, trots att de volymmässigt utgör endast 1–2 procent av hela körteln. Det anses att snabba fluktuationer i dess mikrocirkulation reglerar insulinfrisättningen. När det gäller blodflöde, tillhandahålls arteriell blodförsörjning genom en neurovaskulär stjälk som bär arterioler vilka genom bifurkationer till höggradigt fenestrerade kapillärer förs in i mitten av ön (figur 3.1). Blodkärlen vänder sedan tillbaka mot periferin av öarna, där de somatostatin- och glukagonproducerande D- 2012-02-06 cellerna och alfacellerna är belägna. Genom denna väg är det troligt att parakrint insulin kan påverka utsöndringen av glukagon och somatostatin inom ön. Vid en intravenös bolusinjektion av glukos ökas snabbt – sannolikt via en centralnervös mekanism – blodflödet till de langerhanska öarna vilket är en del i stimuleringen av insulinsekretionen som glukos inducerar (10). Likaså är det visat att etanol utövar en markant effekt på pankreas mikrocirkulation genom att inducera en massiv omfördelning av körtelns blodflöde (11). Etanol ökar markant blodflödet till de langerhanska cellöarna men påverkar inte nämnvärt det totala blodflödet till pankreas, vilket resulterar i en ökad insulinsekretion som inducerar hypoglykemi. Denna mekanism kan potentiera den välkända hämmande effekten av alkohol på leverns glukosproduktion. Figur 3.1 Den langerhanska öns uppbyggnad. 2012-02-06 Denna omfördelning av pankreas blodflöde från den exokrina till den endokrina delen, via mekanismer medierade av NO och vagala impulser (11), kan ligga bakom den välkända hypoglykemiska effekten av alkohol hos diabetiker och hos alkoholister med nedsatt leverfunktion. Denna mekanism kan också vara relevant för den derangerade metabola situationen hos individer med diabetes. Alkohol kan orsaka kraftig och långdragen hypoglykemi hos patienter som behandlas med sulfonylureapreparat, till exempel glibenklamid, eftersom många av dessa läkemedel har en lång biologisk halveringstid. Många alkoholister är dessutom undernärda och/eller har levercirros och kan därför ha svårigheter att mobilisera glukoneogenes som svar på hypoglykemi. Neural kontroll av insulinsekretionen De langerhanska öarna är också till stor del styrda av det autonoma nervsystemet, både sympatikoadrenerga (splankniska) och parasympatikokolinerga (vagala) nervfibrer omsluter öarna (figur 3.1). Det verkar som om båda typerna av nervtrådar inte bara innerverar intrapankreatiska blodkärl, utan också kan sluta i intrapankreatiska ganglier där frisatta transmittorsubstanser lokalt kan modulera öcellernas funktion och hormonsekretion (6). Sympatisk nervstimulering resulterar i hämning av insulin- och somatostatinsekretion, medan däremot glukagonfrisättningen stimuleras. Däremot medför aktivering av vagusnerven, vilket stimulerar parasympatiska nerver, en förstärkt utsöndring av både insulin och glukagon, medan effekten på somatostatin är variabel (6). De intracellulära signalsystem som aktiveras i betacellen på kolinerg stimulering innebär aktivering av fosfolipas C. Fenylefrin (en alfa-1-adrenerg agonist), klonidin (en alfa-2-adrenerg agonist) och adrenoceptorantagonisten propranolol var alla potenta hämmare in vitro av öcellernas proliferation, medan fenylefrin och klonidin även undertryckte den långsiktiga insulinsekretionen (6). Dessa fynd ger 2012-02-06 ytterligare tilltro till den föreslagna rollen av adrenerg stimulering som en hämmare av insulinfrisättningen (6). På senare år har det blivit alltmer uppenbart att ytterligare mekanismer, som ett komplement till de klassiska adrenerga och kolinerga systemen, är operativa i den neurala kontrollen av öcellernas hormonsekretion. Öarna innerveras också av peptiderga nerver, vilka frisätter neuropeptider som direkt påverkar graden av hormonutsöndring (6). Denna 29 aminosyror långa peptid har påvisats i intrapankreatiska nerver hos flera arter och frisläpps vid sympatisk nervstimulering (6). Liksom en annan neurotransmittor som hämmar insulinsekretionen, neuropeptid Y (NPY), har galanin lokaliserats till intrapankreatiska sympatiska ganglier och därför föreslagits fungera som en sympatisk signalsubstans (6). Receptorer med hög affinitet för galanin har identifierats på betaceller, vars bindning till galanin medför hämmad insulinsekretion från cellen (6). De mekanismer genom vilka galanin försämrar insulinsekretionen är emellertid inte helt klarlagda. Det har dock konstaterats att galanin inducerar en snabb minskning av betacellens membranpotential och cytoplasmiska fria Ca2+koncentration, vilket tyder på att neuropeptiden inducerar repolarisering i betacellen genom ökad K+-konduktans (6). Ytterligare mekanismer för galaninets minskning av insulinfrisättningen kan innefatta direkt hämning av exocytosprocessen, eftersom galanin också hämmar insulinfrisättningen från permeabiliserade betaceller på ett Ca2+-oberoende sätt (6). Dessutom verkar det som om effekten av galanin förmedlas genom medverkan av ett hämmande GTP-bindande protein, eftersom dess effekter motverkas av förbehandling med pertussistoxin (6). Det finns även neuropeptider som kan stimulera insulinfrisättningen. Dessa inkluderar vasoaktiv intestinal polypeptid (VIP), pituitär adenylatcyklasaktiverande polypeptid (PACAP) och gastrinfrisättande peptid (GRP), som alla frisätts i pankreas vid aktivering av vagusnerven och även främjar frisättning av insulin och glukagon in vitro (6). 2012-02-06 Inkretinsystemet Det har länge varit känt att insulinfrisättningen blir avsevärt större om glukos ges peroralt istället för intravenöst, trots att samma blodsockerstegring sker. Detta är den så kallade inkretineffekten och antyder att det måste finnas något reglersystem i gastrointestinalkanalen som kan potentiera glukosstimulerad insulinfrisättning. Faktum är att man redan 1906 började ana existensen av en sådan faktor då man, långt innan insulinets upptäckt, lyckades minska graden av glukosuri hos patienter med typ 1-diabetes genom att oralt ge dem ett extrakt av tunntarmsslemhinna (12). Idag vet vi att det i tunntarmen, liksom i resten av gastrointestinalkanalen, finns enteroendokrina L-celler. Dessa producerar inkretinhormoner, bland annat glukagonlik peptid 1 (GLP-1) och glukosinsulinotropisk polypeptid (GIP). Sekretionen av inkretinhormonerna stimuleras av födointag, bland annat glukos, och har till uppgift att dels stimulera insulinsekretionen från pankreas, dels hämma glukagonfrisättningen (13). Dessa två mekanismer, jämte flera andra, bidrar till att förbättra glukoshomeostasen vid T2D som ju karakteriseras av en otillräcklig nivå av insulin (vilket medför defekt glukosupptag i framför allt skelettmuskulatur) men även en oproportionerlig ökning av glukagon (som ökar leverns glukosproduktion). GLP-1 är idag basen för den senaste klassen av antidiabetiska läkemedel, inkretinbaserad terapi, som berörs på annat håll i denna bok. Till skillnad från andra läkemedel som stimulerar insulinfrisättningen, till exempel sulfonylureapreparat, är både den ökade insulinfrisättningen och den hämmade glukagonsekretionen beroende av glukos. Båda effekterna är uttalade vid högt glukos (när det behövs) men upphör när normoglykemi inträder. Således finns här, till skillnad mot sulfonylureapreparat, två inbyggda säkerhetsventiler mot hypoglykemi och viktuppgång som båda kan orsakas av hyperinsulinemi. Det är visat i djurförsök samt ex vivo i humana betaceller att GLP-1 och dess analoger, förutom ovanstående akuta effekter, även verkar kunna utöva trofiska effekter på betacellerna i så måtto att betacellernas proliferation ökas och deras apoptotiska celldöd (som tros vara orsakad av diabetisk 2012-02-06 glukolipotoxicitet) minskas. Detta är mycket spännande effekter som, om de kan reproduceras i T2D-patienter, kan tänkas utgöra en sjukdomsmodifierande mekanism genom att skydda och stimulera betacellen. Huruvida detta kan bryta det progressiva naturalförloppet av T2D, som ju karakteriseras av en gradvis förlust av betaceller, återstår att se. Pulsatil insulinsekretion Insulin frisätts inte konstant utan i ett oscillerande mönster, även under fasta, hos friska individer (14). Detta scenario är inte ovanligt inom endokrinologin: kardinalexemplet är gonadotropinerna som hos kvinnor uppvisar månadsvisa fluktuationer, men även hypofyshormoner och glukokortikoider företer ju betydande variationer över dygnet (”cirkadiska rytmer”). Att glukosnivån i blod hos friska oscillerar även i fasta är dock ingen ny kunskap, faktum är att de första rapporterna om detta kom 1923, det vill säga redan året efter insulinets upptäckt! I takt med att antikroppsbaserade diagnostika för insulin utvecklades, kunde man konstatera att även insulinnivåerna i blod hos friska djur och människor oscillerar med olika periodicitet och att hos friska individer cirka 75 procent av den totala insulinsekretionen sker pulsatilt (14). Sedermera visades att även motreglerande pankreashormon som glukagon oscillerar, men antisynkront med insulin och glukos (14). Det är heller inte enbart glukos som styr pulsatiliteten under fysiologiska förhållanden, till exempel har fria fettsyror – viktiga substrat i betacellens energimetabolism – likartade effekter (14). Oscillerande biologiska funktioner är ett globalt fenomen som inte alls är begränsat till insulin och andra hormoner, utan flertalet inter- och intracellulära signalsystem (såsom Ca2+, cykliskt AMP, intermediärmetaboliter med mera) i de flesta organ i kroppen uppvisar oscillerande fluktuationer (15). 2012-02-06 Bortfall av pulsatil insulinfrisättning är en tidig defekt vid typ 2-diabetes Vad är då nyttan med att frisätta insulin pulsatilt och har störningar i detta något med diabetes att göra? Eftersom pankreas dräneras venöst i leverns portåder (vena porta), har levern en nyckelroll i insulinhanteringen och kanske inte helt förvånande sker även leverns glukosproduktion pulsatilt. Insulinnivåerna i portablod är betydligt högre än i perifert venblod, vilket beror på att levern extraherar 50–80 procent av insulinet i dess förstapassage genom organet och på utspädningseffekten i den betydligt större systemiska blodvolymen, och uppvisar också avsevärt större svängningar (flera hundra gånger) jämfört med i perifert venblod (~ 3–5 gånger [14, 15]). Eftersom insulinreceptorn internaliseras i cellen då det bundit till insulin, är en attraktiv hypotes att pulsatil insulinsekretion har som ändamål att säkerställa adekvat insulinreceptorförekomst. Annorlunda uttryckt: pulsatil insulinfrisättning garanterar att insulinreceptorerna uppregleras vid låga insulinnivåer, en mekanism som således kan tjäna till att förhindra uppkomst av insulinresistens. Till stöd för denna hypotes har noterats att det krävs mindre mängd insulin för att upprätthålla normoglykemi om hormonet ges i pulser än om motsvarande mängd ges som konstant infusion, ett fenomen som kunnat tillskrivas högre uttryck av insulinreceptorer (16). Den pulsatila insulinfrisättningen är störd vid diabetes typ 2, även mycket tidigt i sjukdomsförloppet, och det förefaller som om det framför allt är amplituden i oscillationerna, och inte frekvensen, som är derangerad. Faktum är att redan förstagradssläktingar till patienter med typ 2-diabetes uppvisar oregelbundenheter i och bortfall av pulsatil insulinsekretion trots att deras glukostolerans endast är minimalt påverkad, vilket antyder att detta kan vara en mycket tidig och därmed också patogenetiskt betydelsefull defekt i utvecklingen av typ 2-diabetes (17). Likaså förefaller det som om defekten är specifik för glukos, eftersom insulinsvaret på L-arginin var intakt (14). 2012-02-06 Vissa antidiabetiska läkemedel ökar insulinoscillationerna Vissa intracellulära budbärare i betacellen, till exempel Ca2+, cykliskt AMP, oxygenkonsumtion, ATP, polyfosfoinositider, glukosmetaboliter med flera, oscillerar också och utgör således potentiella läkemedelsmål mot diabetes (15). För att ytterligare närma sig den kliniska verkligheten, är det speciellt intressant att notera att inkretiner (GLP-1-baserade substanser) och andra antidiabetiska läkemedel har stimulerande effekter på pulsatil sekretion (14). Kännedom om att verkningsmekanismen för hur inkretinerna utövar sin insulinfrisättande effekt omfattar cykliskt AMP skapar också större förtroende för läkemedelsklassen. Långtidsreglering av insulinsekretionen och defekter vid T2D Det är viktigt att ha klart för sig att det, förutom ovanstående akuta reglermekanismer för kortsiktiga fluktuationer i insulinfrisättningen, även finns långsiktiga förändringar av insulinsekretionen som svar på ökande eller minskande insulinbehov. Exempel på detta är graviditet, fysisk inaktivitet och ökad kroppsvikt (speciellt bukfetma) då insulinresistens gradvis inträder. Hos glukosfriska individer, utan genetisk predisposition, leder detta till att de langerhanska cellöarna genomgår en hyperplasi så att mängden betaceller ökar. Hos genetiskt predisponerade individer sker detta inte i tillräcklig grad, eller pågår bara en kort tid, och dessa individer kommer följaktligen att sakta utveckla glukosintolerans som ofta i förlängningen resulterar i manifest T2D (18). Endokrina pankreas förmåga till plasticitet, det vill säga att långsiktigt anpassa sig till rådande insulinbehov, utgör således en viktig determinant för pankreas totala insulinproducerande kapacitet för att upprätthålla normoglykemi, och defekter i denna plasticitet kan orsaka glukosintolerans och T2D (18). I humana obduktionsmaterial har det visats att den otillräckliga mängden 2012-02-06 betaceller hos patienter med T2D till stor del betingas av ökad apoptotisk celldöd (18). Mekanismerna för denna apoptos är föremål för omfattande forskning, men det anses att den glukotoxicitet och lipotoxicitet som karakteriserar T2D kan medföra funktionshämning och apoptotisk död hos betacellerna. Sannolikt är processer som oxidativ och nitrosativ stress inblandade i detta. Ackumulerade data tyder också på att stress i det endoplasmatiska retiklet (ER) sker i betaceller vid T2D (3). Fria fettsyror kan orsaka ER-stress och tros vara förmedlare av betacellsdysfunktion och apoptotisk död. Likaså är det visat att sulfonylureapreparat orsakar samma skador på humana betaceller ex vivo (19). Huruvida denna glukolipotoxicitet, och/eller deletära effekter av sulfonylureapreparat, i klinisk praxis reflekteras av diabetessjukdomens progressivitet och tablettsvikt återstår emellertid att leda i bevis. Förutom denna toxicitet är det sannolikt att även amyloidinlagring i öarna spelar en klar roll för minskad betacellsfunktion vid T2D. Bland de faktorer som stimulerar betacellens proliferation kan nämnas glukos, GH, gastrin, insulin, IGF-1, GLP-1 med flera (20). Utrymmet i denna bok tillåter dessvärre inte någon detaljerad mekanistisk redogörelse för de intracellulära signalvägar som kopplar ett tillväxtstimulus (till exempel GLP-1) till mitotisk delning av betacellen utan den intresserade läsaren hänvisas till översiktsartiklar i ämnet (21, 22). Replikering av betacellerna är en viktig källa till betacellens expansion i tidig barndom. Den senaste tidens koppling mellan T2D och flera transkriptionsfaktorer inblandade i cellcykeln har lett till teorin att tillväxten av betacellsmassan i den tidiga barndomen kan vara en viktig faktor för risken att utveckla T2D i vuxen ålder (21). Även om det finns ett stort intresse för att hitta sätt att stimulera betacellerna till proliferation (antingen endogena betaceller in vivo eller exogena ex vivo inför transplantation) för att kunna återställa bristen på betaceller vid T2D, så måste man hålla i minnet att replikerande betaceller (liksom flertalet andra celltyper) har en ökad sårbarhet för apoptos. Detta kommer sannolikt att begränsa det terapeutiska värdet hos stimulering av betacellsreplikering i den 2012-02-06 proapoptotiska miljön vid T2D, såvida denna inte görs samtidigt med en strategi för att undertrycka ökad apoptos (till exempel med GLP-1, som även stimulerar prekursorceller i pankreas duktepitel till att differentieras ut till betaceller). Utvecklingen av läkemedel som reglerar dessa parametrar kommer att bli en stor utmaning för akademi och industri under de kommande åren i behandlingen av T2D. Nedsättningen i betacellsfunktion och eventuellt också betacellsmassan verkar vara reversibel, särskilt vid tidiga stadier av sjukdomen där den begränsande tröskeln för reversibilitet ännu inte nåtts. Bland insatserna för att bevara eller ”föryngra” betaceller kan nämnas kortsiktig intensiv insulinbehandling av nydiagnostiserad T2D som kommer att öka betacellsfunktionen, sannolikt genom att avlasta den endogena insulinproduktionen så att betacellen får vila. En annan intervention för att inducera betacellsvila kan ske genom selektiv aktivering av K ATP -kanaler med hjälp av droger som diazoxid. En tredje typ av intervention är användningen av apoptoshämmande läkemedel, såsom tiazolidindioner (TZD) och inkretinbaserad terapi (till exempel GLP-1-analoger) som båda har visat signifikanta kliniska tecken på positiva effekter på människors betacellsfunktion. TZD har indirekta effekter på betacellsfunktionen genom att insulinkänsligheten ökas, så att trycket på betacellerna att överproducera insulin minskar. De direkta effekterna av TZD sker via peroxisomproliferatoraktiverad gammaaktivering (PPAR-gammaaktivering) i de langerhanska öarna vilket, tillsammans med ovanstående indirekta effekter, leder till förbättrad betacellsfunktion (23). Självklart är det svårt att uppskatta de skyddande effekterna av TZD och inkretiner på betaceller hos människor, och det finns idag inga robusta kliniska bevis på att dessa läkemedel verkligen har skyddande effekt på humana betaceller. Framtidsperspektiv Regleringen av stimulussekretionskopplingen i pankreas insulinproducerande betaceller har varit föremål för massiva 2012-02-06 forskningsinsatser under många år, men trots att snart 100 år gått sedan insulinets upptäckt har man ännu inte lyckats lokalisera de defekter i betacellen som kan orsaka typ 2-diabetes. På senare år har emellertid nya kontrollmekanismer för insulinfrisättningen identifierats, med potentiell relevans för uppkomsten och behandlingen av typ 2-diabetes. Beträffande mål för framtida läkemedel som stimulerar insulinsekretionen kan nämnas (förutom aktivatorer av glukokinas [se ovan]) agonister till Gproteinkopplad receptor 119 (GPR119) som visats stimulera inte bara insulinfrisättning från betacellen, utan också sekretionen av GLP-1 från tunntarmens enteroendokrina L-celler (24). Ett flertal företag testar i tidig fas GPR119-agonister som således har potential att korrigera två hormonella störningar vid T2D, nämligen brist på insulin och GLP-1. Referenser 1. U.K. Prospective Diabetes Study Group. U.K. prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. Diabetes 1995;44(11):1249–1258. 2. Fred RG, Welsh N. The importance of RNA binding proteins in preproinsulin mRNA stability. Mol Cell Endocrinol 2009;297(1–2):28– 33. 3. Ortsäter H, Sjöholm Å. A busy cell – endoplasmic reticulum stress in the pancreatic β-cell. Mol Cell Endocrinol 2007;277(1–2):1–5. 4. Kahn SE, Halban PA. Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes 1997;46(11):1725–1732. 5. Luppi P, Cifarelli V, Wahren J. C-peptide and long-term complications of diabetes. Pediatr Diabetes 2011;12(3 Pt 2):276–292. 6. Sjöholm Å, Efendić S. Neurohormonal regulation of the insulin stimulussecretion coupling in pancreatic islets. Exp Clin Endocrinol Diabetes 2001;109 Suppl 2:S109–121. 7. Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov 2009;8(5):399–416. 2012-02-06 8. Gembal M, Gilon P, Henquin JC. Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J Clin Invest 1992;89(4):1288–1295. 9. Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in β-cell function. Nature 2001;414(6865):788–791. 10. Jansson L, Hellerström C. Glucose-induced changes in pancreatic islet blood flow mediated by central nervous system. Am J Physiol 1986;251(6 Pt 1):E644–647. 11. Huang Z, Sjöholm A. Ethanol acutely stimulates islet blood flow, amplifies insulin secretion, and induces hypoglycemia via nitric oxide and vagally mediated mechanisms. Endocrinology 2008;149(1):232–236. 12. Moore B. On the treatment of Diabetus mellitus by acid extract of Duodenal Mucous Membrane. Biochem J 1906;1(1):28–38. 13. Holst JJ, Vilsbøll T, Deacon CF. The incretin system and its role in type 2 diabetes mellitus. Mol Cell Endocrinol 2009;297(1–2):127–136. 14. Pørksen N. The in vivo regulation of pulsatile insulin secretion. Diabetologia 2002;45(1):3–20. 15. Tengholm A, Gylfe E. Oscillatory control of insulin secretion. Mol Cell Endocrinol 2009;297(1–2):58–72. 16. Matthews DR, Naylor BA, Jones RG, Ward GM, Turner RC. Pulsatile insulin has greater hypoglycemic effect than continuous delivery. Diabetes 1983;32(7):617–621. 17. O'Rahilly S, Turner RC, Matthews DR. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med 1988;318(19):1225–1230. 18. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. βcell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003;52(1):102–110. 19. Maedler K, Carr RD, Bosco D, Zuellig RA, Berney T, Donath MY. Sulfonylurea induced β-cell apoptosis in cultured human islets. J Clin Endocrinol Metab 2005;90(1):501–506. 2012-02-06 20. Sjöholm Å. Glucose stimulates islet β-cell mitogenesis through GTPbinding proteins and by protein kinase C-dependent mechanisms. Diabetes 1997;46(7):1141–1147. 21. Lee YC, Nielsen JH. Regulation of β-cell replication. Mol Cell Endocrinol 2009;297(1–2):18–27. 22. Sjöholm Å. Diabetes mellitus and impaired pancreatic β-cell proliferation. J Intern Med 1996;239(3):211–220. 23. Zeender E, Maedler K, Bosco D, Berney T, Donath MY, Halban PA. Pioglitazone and sodium salicylate protect human β-cells against apoptosis and impaired function induced by glucose and interleukin-1β. J Clin Endocrinol Metab 2004;89(10):5059–5066. 24. Jones RM, Leonard JN, Buzard DJ, Lehmann J. GPR119 agonists for the treatment of type 2 diabetes. Expert Opin Ther Pat 2009;19(10):1339–1359.