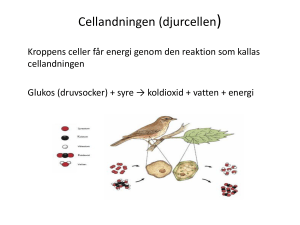
Biomedicinarlinjen, läsår 3
advertisement

Laborationer i Cell- och molekylärbiologi Lab 5-7 Biomedicinprogrammet, termin 2 VT 2014 Laborationerna 5-7 för kursen i Cell- och molekylärbiologi VT 2014 (Biomedicinprogrammet, termin 2) Lokal: Cellodlingslab, C6:122b om ej annat anges Innehållsförteckning Sidan Ordnings- och säkerhetsföreskrifter 3 Röda tråden 4 LAB 5: Sterilteknik, upptining och odling av COS-celler 5 LAB 6A: Studier av intracellulär proteintransport med hjälp av fusionsproteiner med eGFP i COS-celler 9 LAB 6B: Studie av apoptos med hjälp av fusionsprotein med eGFP i COS-celler 14 LAB 7: Kärnfärgning med bisbenzimide (Hoechst 33258) 19 2 Ordnings- och säkerhetsföreskrifter 1. 2. 3. 4. 5. På laboratoriet skall (rentvättad) skyddsrock användas. Rökning, ätande eller dylikt får inte förekomma på laboratoriet. Gäller även snusning. Handtvättning skall ske före och efter laborationen. Före och efter arbetet spritas LAF/sterilbänken av. Betrakta använt material som potentiell smittorisk. Behandla cellerna som om de vore sjukdomsframkallande (patogena). Vid spill av cellösning torka torrt med papper och tvätta sedan med sprit. 6. Smittförande avfall (eukaryota celler) kastas i där för avsedda riskkartonger (GUL märkning). Trasigt glas samt stickande avfall läggs i lämplig burk och kastas i riskkartonger (BLÅ märkning). Annat avfall kastas i papperskorgarna. 7. Tag bort märkning från använt glasmaterial och skölj i vatten. 8. Gör dig bekant med brand- och räddningsutrustning som finns vid/i laboratoriet. 9. Informera dig om användandet av ev. hälsovådliga ämnen i laborationerna. 10. Varje person i gruppen skall föra journal över laborationsarbetet. Arbetet på laboratorium samt arbetet med genmodifierade organismer (GMO) är reglerat enligt svensk och europeisk lagstiftning. Vi hänvisar bl.a. till Arbetarskyddsstyrelsens författningssamling AFS1997:10 ”Laboratoriearbete med kemikalier” samt AFS 2000:5 ”Innesluten användning av genetiskt modifierade mikroorganismer”. Riskbedömning av arbetet samt skadeförebyggande åtgärder skall genomföras innan arbetet påbörjas. Vid frågor eller oklarheter kring arbetets risker skall du tala med labamanuenserna. 3 4 LABORATION 5: Sterilteknik, upptining och odling av COS-celler Syfte: Att lära sig odla celler och studera dessa i mikroskop. Beskrivning: Tina ampuller med COS-celler enligt bifogade anvisningar. Sköt om cellerna regelbundet. COS-celler är en fibroblastcellinje som ursprungligen kommer från njuren hos den gröna markattan (Cercopithecus aethiops). Dessa celler har ett förändrat genom (COS = CV-1, Origin, SV40) som ger uttryck av ett protein, det s.k. stora T-antigenet från SV-40 virus. Tantigenet behövs för att replikation skall kunna ske av vektorer som bär SV40 ori. På grund av det förändrade genomet skyddsklassas arbete med cellerna som arbete med genetiskt modifierade organismer (GMO) av typ II. Detta kräver skyddsbänk av den typ som finns på kurslab och att material inte sprids med vanligt avfall. Gör sammanställning över dels hur många dagar cellerna behövde för att hämta sig efter upptiningen (morfologi), dels hur snabbt de växte sig täta (uppskatta tätheten i % av bottenytan i odlingsskålen varje dag efter upptiningen, plotta en tillväxtkurva)! Lämnas in med labrapport 6! Exempel tillväxtkurva 100 80 60 % täthet 40 20 0 Dag 1 Dag 2 Dag 3 5 Dag 4 Dag 5 UPPTINING AV CELLER Tag upp ett kryorör med celler ur flytande kväve. Tina cellröret i 37°C vattenbad. Tvätta utsidan av röret med 70% alkohol, låt röret lufttorka. Överför cellerna till kulturmedium i ett rör med 5 ml DMEM+ 10% HI FCS. Centrifugera cellerna vid 1000 rpm i 5 min. och resuspendera cellpelleten i 5 ml medium och sätt ut suspensionen i cellodlingsplatta. Inkubera cellerna i inkubatorn. Material 5ml odlingsskålar Centrifugrör Glaspipetter Pasteurpipetter DMEM 10%FCS För din information lämnas här en beskrivning hur man fryser in celler (utförs inte på denna kurs). Därefter beskrivs hur man subkultiverar celler, kallas även splitta, dela eller passera celler. Detta behöver utföras för att cellerna du odlar skall må bra samt för att expandera och få fler celler. INFRYSNING AV CELLER Cellerna bör frysas i tillväxtfas (vid en täthet på ca 6 x 106 celler/ml). Skörda cellerna som skall frysas. Räkna cellerna och centrifugera dessa vid 1000 rpm under 5 min. Tag under tiden fram ett isbad och ställ ner kylt DMSO-medium. När cellerna snurrat färdigt; sug av supernatanten och tillsätt DMSO-mediet för önskad täthet (dvs till ca 106-107 celler/ml). Cellsuspensionen pipetteras upp i 2 ml kryorör, märkta med celltyp, celltäthet och datum. Rören fästs i kanisterstickor och sätts i -70°C under 2 timmar eller över natt. Stickorna flyttas sedan till en kanister med flytande kväve. Gör protokoll över nedfrysta celler, anteckna kanisternummer. DMSO-medium 80 ml DMEM (komplett Dulbecco's minimalmedium) 10 ml DMSO (dimetylsulfoxid), toxiskt! 10 ml värmeinaktiverat kalvserum (FCS= fetal calf serum, HI = heat inactivated) DMSO kan inte sterilfiltreras eller autoklaveras men kan anses sterilt. 6 SUBKULTIVERING AV CELLER Introduktion När normala eukaryota celler får växa i odlingskärl, t ex en petriskål, bildar de till slut ett jämnt lager över hela ytan. Kulturen sägs då vara konfluent. Celldelningen avstannar p g a kontaktinhibering. För att cellerna skall kunna växa vidare måste de spädas och detta sker vid subkultivering då kulturen delas och förs vidare till nya skålar. Denna procedur kan bara upprepas ett visst antal gånger beroende på celltyp innan cellerna dör. Ibland sker mutationer så att cellerna kan fortsätta att växa under obegränsad tid utan att de dör vilket innebär att de kan subkultiveras ett oändligt antal gånger. Cellerna har blivit immortaliserade d v s en etablerad cellinje har skapats. Detta sker lätt spontant hos celler från råtta och mus men aldrig från människa. De immortaliserade cellerna kan förändras ytterligare t ex förlora kontaktinhiberingen. Dessa celler har då blivit transformerade och motsvaras av tumörceller in vivo. Vid subkultivering används trypsin och/eller en EDTA-lösning för att lösgöra cellerna från underlaget. Trypsin klyver proteiner på cellernas yta som deltar i celladhesionen och EDTA binder metalljonerna Mg2+ och Ca2+, vilket medför att celladhesionsproteiner bl.a. cadheriner som är beroende av dessa joner förlorar sin aktivitet. Efter tillsats av trypsin och/eller EDTA genomgår cellerna en morfologisk förändring. Från att vara utsträckta mot underlaget och ganska platta och med synliga stressfiberbuntar så börjar cellerna bli sfäriska och tappa kontakten med underlaget. När cellodlingsskålarna blivit heltäckande med celler = 100% konfluens, delas de vanligen 1:2 eller 1:3. Kontrollera regelbundet cellerna i inverterat faskontrastmikroskop dagarna efter upptining eller senare (ca var 3:e dag). Material DMEM 10% FCS 0,25% trypsin Odlingsskålar Glaspipetter Pasteurpipetter Utförande: 1. Sug av mediet (vakuumsug, steril pipett). 2. Sätt till en liten mängd (2-3 ml) 0,25% trypsinlösning (i kalcium- och magnesiumfri fosfatbuffrad saltlösning = Ca-Mg-fri PBS). Låt cellerna stå i den tunna vätskefilmen 1-2 min i rumstemperatur. Sug därefter av trypsinlösningen (vicka på plattan) men låt en liten mängd vara kvar. 3. Sätt in cellerna i inkubatorn (37°C, 5 % CO2). Kontrollera i mikroskopet hur snabbt cellerna släpper från plasten (olika mellan olika cellinjer). 7 4. När cellerna lossat, tag t ex 10 ml DMEM med 10 % FCS samt pipettera upp och ner så att cellerna lossnar helt från odlingsplattorna. Sätt ut cellsuspensionen i 5 ml per 5 cm platta. (cellodlingsplast, OBS! ej bakterieplattor som har annan sorts plastyta). 5. Märk locket med cellinje, (passagenummer), datum och namn eller gruppnummer. 8 LABORATION 6A: Studier av intracellulär proteintransport med hjälp av fusionsproteiner med eGFP i COS-celler Bakgrund: Det är av stor betydelse att kunna få in främmande DNA i celler och därigenom förändra dem. Utan denna metodik skulle dagens utvecklade bioteknik inte kunnat ske. I detta moment skall ni elektroporera in plasmider som kodar för proteinet "green fluorescent protein" (GFP) i COS-celler. Proteinet är grönfluorescerande om det belyses med blått ljus vid fluorescensmikroskopi och kommer ursprungligen från maneten Aequorea victoria. Det naturligt förkommande proteinet har modifierats genetiskt så att fluorescensen har starkare intensitet och ett snävare spektrum. Det modifierade proteinet kallas enhanced GFP (eGFP). eGFP har visats sig vara mycket användbart som biologisk markör. Proteinet behöver inte någon co-faktor eller substrat för att fluorescera. Det är möjligt att använda GFP i levande celler. eGFP används främst sammansatt med något annat protein och eGFP kan då "lifta" med detta andra protein och "skvallrar" om vart det tar vägen i cellen. Man kan på detta sätt följa vart i cellen ett protein transporteras (sorteras, "protein sorting"). I laborationen kommer ni att använda två olika eGFP plasmider. En plasmid som kodar för eGFP som är sammansatt med en peptid som sorteras till kärnan, och en annan som kodar för eGFP som är sammansatt med en peptid som sorteras till cellmembranet. ”Ensamt” är eGFP ett cytoplasmatiskt protein och kommer att hamna fritt i cytoplasman. Protein sorteras till olika delar av cellen med hjälp av "adresslappar" eller signaler som finns på proteinerna. Den vanligaste signalen är signalen för att utsöndra eller för att hamna i membran (membranbundna protein): den sk. signalsekvensen: Endoplasmatiskt retikulum "ER signal sekvens". Denna signal består av 16-30 aminosyrorna i N-terminalen av polypeptiden, varav 6-12 ofta är hydrofoba. Signalen styr in proteinet i endoplasmatiskt retikulum och klyvs sedan av. Membranproteiner sätts in i membranet medan proteiner som skall utsöndras finns i lumen på ER. Proteinerna förs till Golgi-apparaten och transporteras vidare i membranblåsor och utsöndras eller hamnar i tex. plasmamembranet (Lodish Kap. 13.1-2 samt 14). På ett liknande sätt kan proteiner sorteras till kärnan. Cytoplasmatiska proteiner som bär på en kort sekvens som kallas nuclear localisation signal (NLS) känns igen av ett specifikt receptorprotein. Receptorproteinet transporterar alla protein som har en NLS signal in i kärnan (se Lodish kap 13.6). I den här laborationen kommer ni att överföra två olika GFP-plasmider till eukaryota 9 COS-celler med hjälp av ett starkt elektrisk fält, sk. elektroporation (EP). Andra sätt att överföra DNA är med hjälp av DEAE-Dextran eller kalciumfosfatprecipitation. Även lipofila substanser kan utnyttjas för DNA-överföring (t.ex. Lipofektin, Lipofektamin, GenePorter 2, FuGene 6). Dessa typer av överföring kallas transfektion. COS-cellerna är som konstaterades i Lab 5, en fibroblastcellinje med ett förändrat genom (COS = CV-1, Origin, SV40). Uttryck av det stora T-antigenet från SV-40 virus, startar replikationen hos plasmider som innehåller ett SV40-ori. Ett stort antal plasmider innehållande SV40-ori blir resultatet. Trots det stora antalet kopior kommer plasmid-DNA att spädas ut vid upprepade celldelningar och så småningom försvinna. Expressionen kommer då att avta. Detta sker efter c:a 4-6 dagar. Denna övergående expression kallas transient expression till skillnad från stabil expression där man först selekterat för att den tillförda genen inkorporerats i cellens genom. Syfte: Att studera intracellulär proteinstransport med hjälp av eGFP och fluorescensmikroskopi och att visa hur proteinsorteringssignaler påverkar lokaliseringen av proteinuttryckt i COS celler. DNAfragment kodande för GFP med antingen NLS (pEGFP-NLS) eller MEM (pEGFPMEM) som klonats in i MCS i plasmidvektorer. pEGFP plasmiderna består av: CMV IE; cytomegalovirus promotor, stark viruspromotor som driver expressionen av GFP. Avslutas med SV40 polyA signal. SV40 ori; för replikation i COS celler. Neoresistensgenkassett för eukaryoter: SV40 promotor driver neo-genen. Kassetten ger också möjlighet till selektion i bakterier (kanamycin) dock från annan bakteriepromotor. NLS; nuclear localization signal. MEM; signaler för att tas upp i ER samt skickas till plasmamembranet. 10 Dag 1 Material och utförande: se sid 7: subkultivering av COS-celler. 2 COS plattor görs i ordning med lagom celldensitet så att de får 90-100% konfluens till nästföljande laborationsdag (Dag 2). Spara även lite av cellerna som sätts ut på täckglas i en 5 ml platta för kärnfärgning med Hoechst (s. 19). Dag 2, Elektroporation Material Plasmid DNA: pEGFP-NLS pEGFP-MEM 2 COS-cellsplattor 0,25% trypsin (sorteras till kärnan) 1.0 µg/ elektroporation (sorteras till membranet) 1.0 µg/ elektroporation DMEM+10% FCS PBS 2 st EP kyvetter Utförande: DNA överförs till COS-celler i denna laboration med hjälp av ett starkt elektrisk fält, elektroporation (EP). Ställ in parametrarna vid elektroporationen till 380V, 250µF. Tillsätt rätt mängd plasmid-DNA till respektive elektroporation: 1µg pEGFP-NLS, 1µg pEGFP-MEM. Efter EP, sätts cellerna ut i var sin 5ml platta med DMEM+10% FCS och inkuberas i CO2 inkubator vid 37°C. Bästa resultat erhålles om cellerna vid analysen håller c:a 50% konfluens. 11 ELEKTROPORATION AV COS-CELLER Material EP-buffert = PBS Elektroporationskyvetter 0,25% trypsin DMEM 10% FCS Plasmid pEGFP-NLS Plasmid pEGFP-MEM 2 plattor COS-celler per grupp Bürkerkammare Centrifugrör Cellodlingsplattor 5 ml Glaspipetter Pasteurpipetter Pipetter + spetsar 1. Låt cellerna på era 2 plattor växa till minst 90% konfluens. 2. Sug av mediet och sätt till 0,25% trypsinlösning ca. 2 ml/platta. Låt stå ca.1 min. Sug av så att en liten mängd vätska finns kvar. Låt stå i 37oC i ca 5 min. 3. Häll på 5 ml Elektroporationsbuffert per platta och dissociera cellerna, sätt cellerna i 2 centrifugrör. Dissociera igen med pasteurpipett och tag en droppe cellsuspension från ett av rören för räkning i Bürkerkammare* (se sista sidan i detta kompendium, Bürkerkammaren är inte steril) och centrifugera ned resten av cellsuspensionerna i varsitt provrör. 4. Sug av supernatanten och lös upp pelleten i 0,6 ml EP-buffert per rör. Sätt till 1g plasmid DNA/prov. Blanda och flytta över till två EP-kyvetter. 5. Ställ in elektroporatorn på 380V, 250µF. Stoppa ner kyvetterna (en i taget) och avge pulserna. 6. Överför de porerade cellerna med en pasteurpipett till 2x 5 ml cellodlingsmedium, som sätts i 2x 5 ml plattor. Titta på cellerna för att se hur de ser ut. 7. Inkubera i 37ºC, 5% CO2 tills det är dags att mikroskopera (Dag 3). Dag 3, Mikroskopering Analys av elektroporerade och kärnfärgade celler med hjälp av fluorescensmikroskopi. Analysen sker på forskningslab A2:2 och assisteras av lärare. 12 Uppgifter: 1. Jämför cellerna som elektroporerats med pEGFP-MEM plasmiden med de som fått pEGFP-NLS. 2a. Förklara för labpartnern varför cellerna ser olika ut. 2b Gör en skiss för hur NLS- respektive MEM-GFP proteinet transporteras i COS-cellerna. 3. Räkna hur många celler som totalt finns och hur många som tagit upp DNA (dvs. som är gröna) för respektive plasmid. Labrapport: Kortfattat om syfte och utförande. Bilägg tillväxtkurvan från Lab 5 Redovisa hur spädningar gick till (plasmidspädning ) och hur ni fick fram antal celler från räkningen i bürkerkammaren. Varför användes COS-celler? Redovisa uppgifterna ovan och bilägg skiss samt foton/bilder på cellerna. Beräkna andelen av cellerna som tagit upp respektive EGFP plasmid. Skiljer andelarna sig åt? Varför kan det skilja? Hur kommer det sig att vissa celler ”lyser” starkt och vissa svagt? Slutsats och eventuella felkällor. Hur går fluorescensmikroskopi till? Jämför Hoechst-infärgningen med mönstret ni fick med pEGFP-NLS. Kan man se kromosomerna under mitosfasen i celler som elektroporerats med pEGFP-NLS? Hur kommer det sig? Inkludera ”del-labrapporten” från kärnfärgningen (s. 21). 13 LABORATION 6B: Studie av apoptos med hjälp av fusionsprotein med eGFP i COS celler Bakgrund Antalet celler i en vävnad kan regleras på i huvudsak två sätt: genom celltillväxt eller celldöd. Celldöden kan ske genom nekros som orsakas av cellskador som stör den osmotiska balansen. Detta ger upphov till Ca2+- och vatten inflöde och metaboliskt kaos i cellen, som sväller upp och frigör lyzosomala enzym som är förödande för cellen. Nekrosen leder ofta sekundärt till en inflammatorisk reaktion som sprider skadorna till närliggande vävnader. Under den embryonala utvecklingen sker en massiv celldöd. Exempelvis så dör hälften av alla neuron som bildas i hjärnan. Celldöden måste ske strikt reglerad i tid och rum och utan inflammatoriska reaktioner. Den här typen av celldöd som initierar ett genetiskt programmerat "dödsmaskineri" kallas därför programmerad celldöd där morfologin skiljer sig från nekrosens och kallas apoptos. Under den programmerade celldöden sker en kondensation och klyvning av kromatinet i cellkärnan till 200 bp fragment. Kärnans membran bryts ner, medan det på plasmamembranet tidigt bildas små utbuktningar. Senare uppstår sk apoptotiska kroppar i cytoplasman, dvs membranomslutna vesiklar av cytoplasma innehållet. Detta innebär en fragmentering av cellen som då krymper, i motsats till nekrosens svällande celler. Funktionen för de apoptotiska kropparna är att innesluta cellens materia i mindre paket som grannceller snabbt kan fagocytera och på så sätt förhindra läckage av inflammatoriska komponenter från den döende cellen. Apoptos kan initieras t.ex. genom att beröva celler tillväxtfaktorer, bindning av ligander till TNF och FAS receptorer, DNA skador, virus etc. Dessa stimuli initierar en kaskad av signaler som slutligen leder till att s.k. caspaser aktiveras. Caspaser är cysteinproteaser som klyver en rad olika substrat i cellen som t.ex. leder till att cytoskelettet bryts ned och att DNA fragmenteras. Apoptossignalen kan dock regleras av protein som tillhör Bcl-2- och IAP (inhibitory of apoptosis protein)- familjerna. I Bcl-2-familjen finns både proteiner som hämmar (ex Bcl-2, Bcl-x, Bcl-w) och förstärker (ex Bax, Bad, Bak) apoptossignalen. Dessa proteiner bildar ofta homo- eller heterodimerer och kvoten mellan hämmare och förstärkare verkar påverka dödsförloppet. 14 Bad är ett protein i Bcl-2-familjen som påskyndar celldöden genom att inhibera antiapoptotiska Bcl-2-familjemedlemmar (ex Bcl-2 och Bcl-x) som i sin tur inhiberar caspaser som verkställer dödsmaskineriet. I den här laborationen elektroporerar vi in DNA för Bad i COS-celler för att se om Bad kan inducera celldöd i dessa celler. DNA som kodar för Bad är hopkopplat med DNA för ”green fluorescent protein” (GFP), vilket gör att cellerna som uttrycker fusionsproteinet GFP-Bad fluorescerar grönt om det belyses med blått ljus i fluorescensmikroskop. I kontrollcellerna elektroporerar vi in en tom GFP-vektor. Vi kommer att kombinera den gröna fluorescensen med kärnfärgning (Hoechst-färgning, se s. 19) och kan på så vis se om de gröna cellerna är döda/döende. I den här laborationen använder vi vektorer där det naturligt förkommande proteinet GFP har modifierats genetiskt så att fluorescensen har starkare intensitet och ett snävare spektrum. Det modifierade proteinet kallas enhanced GFP (eGFP). eGFP har visats sig vara mycket användbart som biologisk markör. Proteinet behöver inte någon co-faktor eller substrat för att fluorescera. Det är möjligt att använda GFP i levande celler. Proteinet används främst sammansatt med något annat protein och eGFP kan då "lifta" med detta andra protein och "skvallrar" om och var proteinet uttrycks i cellen. Genom att elektroporera celler med en ”tom” vektor, d.v.s. en vektor som endast uttrycker GFP kan man utesluta att vektorn i sig har någon effekt på cellerna. I den här laborationen kommer ni att överföra två olika plasmider till eukaryota COS-celler med hjälp av ett starkt elektrisk fält, s.k. elektroporation (EP). Andra sätt att överföra DNA är med hjälp av DEAE-Dextran eller kalciumfosfatprecipitation. Även lipofila substanser kan utnyttjas för DNA-överföring (t.ex. Lipofektin, Lipofektamin, GenePorter 2, FuGene 6). Dessa typer av överföring kallas transfektion. COS-cellerna är som konstaterades i Lab 5, en fibroblastcellinje med ett förändrat genom (COS = CV-1, Origin, SV40). Uttryck av det stora T-antigenet från SV-40 virus, startar replikationen hos plasmider som innehåller ett SV40-ori. Ett stort antal plasmider innehållande SV40-ori blir resultatet. Trots det stora antalet kopior kommer plasmid-DNA att spädas ut vid upprepade celldelningar och så småningom försvinna. Expressionen kommer då att avta. Detta sker efter c:a 4-6 dagar. Denna övergående expression kallas transient expression till skillnad från stabil expression där man först selekterat för att den tillförda genen inkorporerats i cellens genom. Syfte: Att överuttrycka genen Bad i levande COS-celler med hjälp av eGFP uttryckande plasmid och därigenom visa Bads inverkan på celldöd. 15 Till vänster visas eGFP plasmid där DNA fragment för Bad har klonats in i MCS. Till höger visas tom eGFP vektor som vi använder som kontroll i laborationen. pEGFP plasmiden består av: CMV IE; cytomegalovirus promotor, stark viruspromotor som driver expressionen av Bad och GFP (pEGFP). Avslutas med SV40 polyA signal. SV40 ori; för replikation i COS celler. Neo-resistensgenkassett för eukaryoter: SV40 promotor driver neo-genen. Kassetten ger också möjlighet till selektion i bakterier (kanamycin) dock från annan promotor. Dag 1, Subkultivering av COS celler. Material och utförande: se sid 7: subkultivering av COS-celler. 2 COS plattor görs i ordning med lagom celldensitet så att de får 90-100% konfluens till nästföljande laborationsdag (Dag 2). Spara även lite av cellerna som sätts ut på täckglas i en 5 ml platta för kärnfärgning med Hoechst (Dag 3). Dag 2, Elektroporation Material Plasmid DNA: pEGFP-Bad pEGFP 2 COS-cellsplattor 0,25% trypsin DMEM+10% FCS PBS 2 st EP kyvetter (pro-apoptotisk gen) 2.0 µg/ elektroporation (kontrollplasmid) 2.0 µg/ elektroporation 16 Utförande: DNA överförs till COS-celler i denna laboration med hjälp av ett starkt elektrisk fält, elektroporation (EP). Ställ in parametrarna vid elektroporationen till 380V, 250µF. Tillsätt rätt mängd plasmid-DNA till respektive elektroporation: 2 µg pEGFP-Bad, 2 µg pEGFP. Efter EP, sätts cellerna ut på täckglas i var sin 2 ml platta med DMEM+10% FCS och inkuberas i CO2 inkubator vid 37°C. Bästa resultat erhålles om cellerna vid analysen håller c:a 50% konfluens. ELEKTROPORATION AV COS-CELLER Material EP-buffert = PBS Elektroporationskyvetter 0,25% trypsin DMEM 10% FCS Plasmid pEGFP-Bad Plasmid pEGFP 2 plattor COS-celler per grupp Bürkerkammare Centrifugrör Cellodlingsplattor 2 ml Glaspipetter Pasteurpipetter Pipetter + spetsar 1. Låt cellerna på era 2 plattor växa till minst 90% konfluens. 2. Sug av mediet och sätt till 0,25% trypsinlösning ca. 2 ml/platta. Låt stå ca.1 min. Sug av så att en liten mängd vätska finns kvar. Låt stå i 37oC i ca 5 min. 3. Häll på 5 ml Elektroporationsbuffert per platta och dissociera cellerna, sätt cellerna i 2 centrifugrör. Dissociera igen med pasteurpipett och tag en droppe cellsuspension från ett av rören för räkning i Bürkerkammare* (se sista sidan i detta kompendium, Bürkerkammaren är inte steril) och centrifugera ned resten av cellsuspensionerna i varsitt provrör. 4. Sug av supernatanten och lös upp pelleten i 0,6 ml EP-buffert per rör. Sätt till 2 g plasmid DNA/prov. Blanda och flytta över till två EP-kyvetter. 5. Ställ in elektroporatorn på 380V, 250µF. Stoppa ner kyvetterna (en i taget) och avge 17 pulserna. 6. Överför de porerade cellerna med en pasteurpipett till 2x 2 ml cellodlingsmedium, som sätts i 2x 2 ml plattor (med täckglas). Titta på cellerna för att se hur de ser ut. 7. Inkubera i 37ºC, 5% CO2 över natt. Dag 3, Kärnfärgning med bisbenzimid (Hoechst 33258). Material och utförande: se sidan 19: utgå ifrån era två 2 ml odlingsskålar med elektroporerade celler från gårdagen samt 5 ml odlingsskål med (icke-elektroporerade) COSceller från Dag 1. Dag 4 och 5, Mikroskopering Analys av elektroporerade och kärnfärgade celler med hjälp av fluorescensmikroskopi. Analysen sker på forskningslab A2:2 och assisteras av lärare. Uppgifter: 1. Uppskatta (räkna) hur stor andel av cellerna som går igenom apoptos för respektive plasmid. 2. Räkna hur många celler som totalt finns och hur många som tagit upp DNA (d.v.s. som är gröna) för respektive plasmid. Laborationsrapport: Kortfattat om syfte och utförande. Bilägg tillväxtkurvan från lab 5 Redovisa för hur spädningarna gick till (plasmidspädning) och hur ni fick fram antal celler från räkningen i bürkerkammaren. Varför användes COS celler? Redovisa uppgifter ovan. Beräkna hur stor andel av cellerna som tagit upp plasmiderna. Hur går fluorescensmikroskopi till? Slutsats och eventuella felkällor. Var är uttrycket av GFP lokaliserat, i kärnan eller i cytoplasman? Stämmer det ni iakttar med teorin? Hur stor andel (%) av de Bad-GFP respektive pEGFP positiva cellerna har apoptotiskt utseende? Beskriv hur ni avgör om cellerna är döende eller inte. Stämmer era resultat överens med teorin? Inkludera ”del-labrapporten” från kärnfärgningen (s. 21). 18 LABORATION 7: Kärnfärgning med bisbenzimid (Hoechst 33258) i COS-celler Bakgrund Färgning av celler i kultur och i vävnad bygger på att specifika färgämnen binder till (färgar) olika delar eller molekyler i cellen. Färgning av cellkärnor kan göras med färgämnen som binder till DNA. Ofta är sådana färgämnen basiska eller har en molekylstruktur som tillåter att de starkt binder in till DNA-dubbelhelixen (interkalerar med DNAt). Basiska färgämnen binder DNA eftersom DNAt är surt (nukleinsyra). Färgämnet i denna laboration (Hoechst 33258) interkalerar med DNA-dubbelhelixen. Andra kärnfärgande ämnen är DAPI, Propidiumjodid, Ethidimbromid och Acridin Orange. Då kärnfärgning används på celler som delar sig kan kärnans och kromosomernas utseende studeras under mitosens faser. Många cellfärgämnen är fluorescerande ämnen (fluoroforer) och studeras därför med hjälp av fluorescensmikroskopi. Studera Lodish et al Kap. 18.6 för att kunna känna igen mitosfaserna! Syfte med kärnfärgning Eftersom levande COS-celler i kultur delar sig så kommer en del celler att befinna sig i mitosfasen. Genom att färga in kärnorna kommer ni kunna urskilja kromosomerna under mitosens olika faser. Man kan också genom färgning av kärnan urskilja apoptotiska celler där cellkärnans kromatin har kondenserats och klyvts. För er som har gjort laboration 6B och där elektroporerat in Bad i levande celler kan därför genom Hoechst färgning avgöra om de celler som uttrycker Bad har gått in i apoptos eller ej. Kort beskrivning av utförandet COS-celler sås ut på täckglas i odlingsskålar. Cellerna kommer att sätta sig på glasen och börja dela sig. Efter ca ett till två dygn fixeras cellerna och färgas med Hoechst No 33258. Cellerna tvättas och täckglaset som de sitter på monteras med cellerna nedåt på ett objektsglas. De färgade cellerna studeras i mikroskop och ni skall försöka hitta celler som befinner sig i de olika faserna av cell cykeln. Bilder tas med digitalkamera. Hoechstfärgning Material Skål med celler på täckglas (från Dag 1 av Lab 6) PBS 19 4% PFA (4% paraformaldehyd i PBS) OBS! PFA är vådligt. Använd hanskar och kasta avfall i där för avsedd slask. Hoechst No 33258 (1 mg/ml, 100x stock) Pincett Monteringsmedium (PBS:glycerol 50:50) Objektsglas Nagellack Aluminiumfolie bisBenzimid är fluorescerande i blått och interkalerar till DNA i A/T-rika regioner. Har låg cytotoxicitet och skadar därför celler relativt måttligt men är ljuskänsligt och skall därför förvaras mörkt. Utförande: 1. Sug av mediet från skålen med täckglaset. 2. Tvätta cellerna en gång med 1 ml PBS. 3. Tillsätt försiktigt 1 ml 4% PFA (rumstempererad). 4. Fixera vid rumstemperatur (RT) i 10 min. 5. Sug av PFA försiktigt och kasta i slaskflaska! 6. Tvätta försiktigt cellerna en gång med 1-2 ml PBS. 7. Tillsätt 1,5 ml PBS innehållande 10µg/ml Hoechst No 33258 (stam 1mg/ml) 8. Svep in skålen i alu-folie och inkubera RT 5 min. 9. Sug av (kasta i slaskflaska) och tvätta försiktigt med 1-2 ml PBS 3 gånger. 10. Gör i ordning ett objektglas med en liten droppe med monteringsmedium mitt på glaset. Märk objektglaset med gruppnummer. 11. Sug av tvättlösningen och plocka försiktigt upp täckglaset med en pincett. Håll koll på vilken sida av glaset cellerna finns på! Sug bort överskottet av PBS från cellerna genom att föra en pappershandduk mot kanten av glaset. Låt dock ej cellerna torka. 12. Placera täckglaset med cellerna nedåt mitt över droppen med monteringsmedium på 20 oblektglaset. 13. Sug upp eventuellt överskott av monteringsmedium med kanten på en pappershandduk och låt glaset torka några minuter. Fäst och förslut sedan kanten på täckglaset mot objektglaset med nagellack och låt torka innan analys i mikroskop. 14. Förvara glasen i mörker (aluminiumfolie) tills mikroskopering. Mikroskopering Analysen assisteras av lärare, korridor A2:2. Uppgifter: 1. Notera hur den normala COS cellkärnan ser ut under interfas. Notera ev. avvikelser i utseende av interfaskärnor. Vad kan dessa bero på? 2. Identifiera celler som genomgår mitos. 3. Identifiera de olika faserna i mitosen i några celler. 4. Uppskatta (räkna) hur stor andel av cellerna som befinner sig i mitos. Del-laborationsrapport (inkluderas i rapporten för laboration 6) Redovisa resultaten på ovan uppgifter. Bifoga ev. utskrifter. Beräkna dessutom hur lång tid mitosen tar med utgångspunkt från hur stor andel av cellerna som befinner sig i mitos och att hela cellcykeln tar ca. 20 timmar för COS celler. 21 *RÄKNING I HEMOCYTOMETER ELLER S.K. BÜRKERKAMMARE Skiss av hemocytometer I det visade exemplet är det 1.0 mm mellan trippellinjerna. Känner man dessutom till djupet, kan man beräkna volymen. I den förstorade delen av hemocytometern visas vilka celler som ska räknas (öppna cirklar) och vilka som inte ska räknas (svarta cirklar). Celler som rör vid mittlinjen i den översta och i den vänstra trippellinjen räknas, medan celler som rör vid mittlinjen i den nedersta och den högra trippellinjen inte räknas. 1. Täckglaset läggs över hemocytometerns räknekammare, så att det vilar på de två upphöjda kanterna på hemocytometern. 2. En liten del av cellsuspensionen överförs med en pipett till hemocytometern. Sätt pipettens spets i mellanrummet mellan räknekammaren och täckglaset och tryck försiktigt ut suspensionen, så att räknekammaren fylls precis. 3. Med ett 10 x objektiv kommer en ruta i stort sett att motsvara synfältet. Normalt räknas 2 rutor på varje sida av hemocytometern. Om det är svårt att räkna bör provet spädas. 4. Med ledning av figuren, beräkna cellkoncentrationen/ml och redovisa i labbrapport. Observera att alla hemocytomerar inte är identiska (mönster och djup kan variera) men det står oftast angivet på hemocytomern. Glöm inte att räkna med eventuell spädning. 22