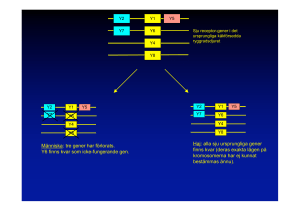
syndrom Prader Willi
advertisement

Sara Cederwall T9‐läkarutbildningen Selective: klinisk genetik. 090105 Prader Willi syndrom Introduktion År 1956 beskrevs för första gången Prader Willi syndromet(PWS). Syndromet beskrevs som en kombination av symptom såsom oförmåga att känna mättnad, fetma, utvecklingsstörning, kortvuxenhet, hypogonadism och neonatal muskelhypotonus. Prader Willi är en neurogenetisk sjukdom som orsakas av en kromosomavvikelse. Alla de nedan beskrivna kromosomavvikelserna leder till undertryck av faderns gener på en viss region (PWS region) på kromosom 15. Skulle det istället vara undertryck av moderns gener, i samma region på kromosom 15, skulle det leda till Angelsmansyndromet(AS).4 Den vanligaste orsaken, ca 60‐70% av fallen, beror på en deletion av kromosom 15, i området 15q11.2‐q13. En del som är nedärvd från fadern. 25‐30% har en dubbel uppsättning av kromosom 15 från modern,(dvs ingen från fadern), en sk maternell disomi. En liten andel av Prader Willi är orsakade av imprintingmutationer. Detta innebär en inaktivering, genom metylering, av en del av kromosom 15 från fadern. En annan liten grupp beror på en translokation, dvs utbyte av genetiskt material, oftast mellan kromosom 12 och 15.4 Även denna kromosomavvikelse leder till avsaknad av arvsmassa från fadern.1 De ända kända ärftliga orsakerna till Prader Willi är just de ovanliga genetiska avvikelserna såsom translokation och imprinting. Det betyder att risken att få ytterligare ett barn med PWS är liten i de flesta fall.4 Prader Willi syndromet är en komplex kromosomavvikelse som involverar flera olika gener. Experter vet fortfarande än idag inte exakt vilka gener eller hur många som är involverade. Därför kan det vara svårt att förklara varför de olika symptomen uppstår. Det är dock känt att generna finns på ett litet område på den långa armen av kromosom 15, området 15q11.2‐13.6 Många av de symptom som patienterna uppvisar tyder på en störning i hypothalamus. Hypothalamus är viktig för reglering av aptit och hungerskänslor, könsutveckling och tillväxt och dessa kan alla vara påverkade vid PW. Det verkar därför troligt att någon signalsubstans saknas eller inte fungerar i detta område.1,4 Varje år föds omkring 6‐8 barn i Sverige i med PWS.1 Symptom Då det inte tillfullo har gått att identifiera den/de gener som är orsaken till Prader Willi är det svårt att förklara varför de olika symptomen uppstår.4 Redan under graviditeten finns det vissa symptom såsom svaga fosterrörelser. Under nyföddhetsperioden är barnet muskelsvagt, har ett svagt skrik och värmeregleringssvårigheter.4 Barnet föds med hypotoni ffa i nacke och bålmuskler. Ett av de tidigaste problemen är födointaget. När barnen föds är de ofta små och underviktiga.1 De nyfödda har dålig sugreflex, som ofta leder till amningssvårigheter. Barnen får ofta sondmatas. Hypotonin är mest uttalad under neonataltiden, men en mild till måttlig hypotoni finns under hela livet. Hypotoni är så karakteristiskt för barn med PWS att en oförklarlig sådan hos ett barn bör föranleda utredning och testning för Prader Willi.2 Under hela spädbarnstiden har barnet problem med matintaget. Det blir succesivt också alltmer märkbart att barnet har en psykomotorisk utvecklingsförsening med särskilda problem med grovmotoriken.4 Under barndomsperioden utvecklar barnet ett karakteristiskt utseende med små mandelformade ögon, smal panna och ett litet ansiktsskelett. Barnets aptit tilltar och undervikt kan snabbt bytas mot övervikt, med en fettansamling ffa på mage och lår. Barnets tillväxt följer inte en normal tilllväxtkurva och en mental utvecklingsförsening blir uppenbar.4 En del barn lider också av skelning och skolios.4 I ca 1‐2 års ålder ändras ätbeteendet och barnen utvecklar en ätstörning med en omåttlig aptit och ett tvångsmässigt förhållande till mat. Dessutom är barnets energibehov lägre än normalt vilket ökar risken ytterligare för att utveckla obesitas. Det ökar även risken för att utveckla diabetes typ 2 (25%av vuxna med PWS har diabetes typ 2)2 samt hjärt och kärlsjukdomar och belastningsskador.1 Minskad mättnadskänsla och hyperfagi är ett uttryck för störningar i hypothalamus. Barnen har också hypogonadism, som leder till underutveckling av de yttre könsorganen och en sen och/eller ofullständig pubertet. Flickorna har ofta en sen menarche eller amenoré. Ytterst lite är studerat om den sexuella aktiviteten, men den ter sig låg och de flesta är infertila. Dock finns bekräftat reproduktion men endast hos kvinnor med syndromet.2 Från Karolinska Institutet är dokumenterat att 3 kvinnor med PWS har fött barn. Det finns inget dokumenterat fall med någon man med PWS som blivit pappa.12 Barnen blir ofta kortväxta med en slutlängd hos kvinnor på 148 cm och hos män 155 cm. Studier talar för att de flesta med PWS har en GH‐brist. Dock är det svårt att fastställa GH‐brist, då feta individer tenderar till att ha ett lågt GH.10 Ofta är barnen ljusare i huden än resten av familjen och har små händer och fötter. Barnen kan ha tuggproblem pga slappa muskler i munhåla och salivet kan vara tjockt, vitt och klibbigt. Detta tillsammans med ätstörningen kan leda till mkt problem med karies. Barnen kan också ha problem med snarkning då de har en låg muskeltonus, samt en anatomi som ger trånga andningsvägar i näshåla och svalg.4 Talutvecklingen är ofta försenad liksom den motoriska utvecklingen att sitta, stå och gå.2 Det tros bero på flera olika faktorer såsom neuromuskulära avvikelser, anatomiska avvikelser i mun och läppar och ofta en utvecklingsstörning,4 Kognitiva funktioner är ofta underutvecklade och begåvningsnivån ligger mellan normalbegåvningens nedre del till svår utvecklingsstörning. Ca 5% av barnen är normalbegåvade och 5% lider av grav utvecklingsstörning. 2,4 Personer med PWS uppvisar också karakteristiska personlighetsdrag som tex känslomässig labilitet och vredesutbrott.2 Många har ett tvångsmässigt beteende, ätstörningen är ett tvång, men även rutinbundenhet och självdestruktiva beteende. Andra tvångsmässiga beteenden som förekommer är ett stort kontrollbehov, överdrivet samlande och de är ofta pedantiska.4 Ca 25% av individer med Prader Willi har autismspektrumstörning. Även ADHD symptom är vanligt i tidig ålder och 5‐10% av individerna har psykoser.2 Många personer med PWS har ett ökat sömnbehov och kan falla i sömn även dagtid. En del har även reagerat oväntat på vissa läkemedel och narkosgaser.1 Den vanligaste orsaken till mortalitet hos PWS är fetmarelaterade kardiovaskulära och respiratoriska sjukdomar hos både vuxna och barn.2 Indikationer för testning av PWS Flera av de ovan nämnda symptomen väcker var och ett för sig eller tillsammans misstanke om PWS. Under neonatalperioden är det ffa en låg muskelhypotonus, dålig sugförmåga och uppfödningssvårigheter som väcker misstanken. Men ju äldre barnet blir desto tydligare blir en kraftig övervikt, psykomotorisk försening, mental retardation, kortvuxenhet och underutvecklade yttre genitalia.17,6 Genetiska varianter av sjukdomen Sjukdomen PWS uppkommer genom ffa tre huvudmekanismer. De olika mekanismerna leder alla till förlust av uttryck av faderns gener inom 15q11.2‐ q13.3 Vid AS undertrycks istället den kromosom som ärvts av modern. Detta visar på att brist på genetiskt material från modern eller fadern ger olika symptombilder.4 Den vanligaste kromosomavvikelsen, vilket innebär ca 60‐70%, är en mikrodeletion av 15q11.2‐q13 regionen på faderns kromosom. 2 Denna region är mycket komplex och de flesta har en av två proximala brytpunket BP1 och BP2 och en känd distal brytpunkt BP3.2 Deletionerna delas in i typ I och II, detta pga av att deletionerna har olika storlek och brytpunkter.6 Typ 1 deletionen involverar BP1(breakpoint1), som ligger nära centromeren. Typ II deletionen involverar BP2 och ligger 500kb distalt om BPI. Båda deletionstyperna slutar i den distala brytpunkten BP3. En typ I deletion ger 500kb mer genetiskt bortfall än en typ II.15 Uniparental disomy (UPD)svarar för ca 25‐30% av PWS. 4 Detta innebär att båda homologerna ärvs från en och samma förälder. Detta kan ske på två sätt, antingen genom fel i meios 2, detta kallas uniparental isodisomy, eller ett fel i meios 1, detta kallas uniparental heterodisomy. Vid PWS har personen nedärvt två kromosom 15 från modern.3 En mindre andel med syndromet, ca 2‐5%, har en inaktivering av en del av kromosom 15, som ärvts av fadern.1,2,4Detta kallas imprintingdefekt och sker via metylering av området.1 Imprintingdefekt innebär att gener i PWSregionen från fadern inte uttrycks som de ska. De flesta imprintingdefekter är epigenetiska och inga DNA sekvensförändringar är funna. De tros uppkomma slumpmässigt under spermatogenesen. Ca 10‐15% av de med imprintingdefeketer orsakas istället av en mikrodeletion i PWS imprintingcenterregionen.2 Även en translokation med en deletion, hos avkomman, som följd kan orsaka PWS.2 Då sker ett utbyte av genetiskt material mellan två kromosomer, oftast mellan 12 och 15. Arvsmassa från faderns kromosom 15 saknas då.1,4 Ett flertal olika gener är identifierade i PWS/AS regionen, men den specifika orsaken till PWS är okänd. Fem gener från den manliga kromosomen är identifierade: SNURF­SNRPN, MKRN3,MAGEL2 och NDN. SNRPN(small nuclear ribonucleoprotein N) är den bäst beskrivna genen, som anses vara involverad i PWS.14 PWS var den först upptäckta sjukdomen som var relaterad till genomisk imprinting och den första som visade sig orsakas av UPD.2 Genetisk‐kliniska varianter En del studier har gjorts på de två vanligaste genetiska avvikelserna dvs UPD och mikrodeletion. I dessa studier har det visat sig att barn med disomi har en lindrigare grad av ”skin picking”, mindre artikulationssvårigheter, ej lika tydliga ansiktsdeformiteter och inte heller är lika uttalat bleka i hud och hår.29 De har en något högre verbal begåvning, men de är sämre på att lägga pussel.4 De har även visat sig ha mindre beteendeproblem. Dock förekommer psykoser och autismspektrum störningar i högre frekvens hos individer med UPD.2 Ny forskning tyder på att begåvningsutvecklingen hänger samman med typen av kromosomavvikelse.4 En studie visade på signifikanta skillnader mellan de två typerna av deletioner. De med typ I visade sig ha sämre läsförståelse och matematik örståelse. De hade också mer beteendestörningar och psykiska problem än de med typ II.16 I studien användes också Woddcock‐Johnson Psycho‐educational Battery, som visade att de med en typ I deletion hade en lägre begåvningsnivå än de med en typ II deletion eller en UPD. Och idag finns identifierat 4 gener mellan BP1 och BP2 där man i musembryon ser att en av generna är uttryckt i mus hjärnan och förmodligen behövs för hjärnans utveckling. Detta kan möjligen förklara varför de med en typ 1 deletion har en lägre begåvningsnivå än de med en typ II deletion.16 I samma studie användes även Wechsler Intelligence Scale och där noterades en signifikant lägre begåvningsnivå hos både de med en typ I och typ II deletion i jämförelse med de med en UPD. Studien visar även på mer självskadebeteende vid deletioner än vid UPD.16 Ärftlighet Risken att få ett barn med PWS är 1/15000.13 Ärftligheten beror på vilken genetisk mekanism som ligger till grunden för sjukdomen. Tidigare i arbetet beskrevs de tre huvudmekanismerna för Prader Willi syndromet. Det är vanligast att orsaken är en nymutation, dvs att sjukdomen uppträder för första gången hos en person och därmed inte är nedärvd. Risken att då få ännu ett barn med PWS är ytterst liten, mindre än 1%. Både vid deletion och UPD är risken mindre än 1%. Ärftliga fall förekommer dock, men är ovanliga. Det har förekommit vid translokation och vid imprintingmutationer.1De flesta med en imprinting defekt har en epigenetisk mutation och risken är då mindre än 1%. Men om mikrodeletionen sitter i imprinting centrat är det risk för en familjär nedärvning och risken kan då vara upptill 50%.2 Om en translokation finns hos någon av föräldrarna kan risken vara upp till 25%. Om det är en de novo translokation är risken mindre än 1%.14 Rådgivning Risken att få ännu ett barn med PWS beror på vilken kromosomavvikelse det första barnet har. En deletion på 15q11.2‐13 är vanligtvis sporadisk och risken är mindre än 1%.11 Dock kan en deletion bero på en translokation och risken kan då vara ökad. 14 Dessa föräldrar och familjer skall erbjudas fortsatt utredning och rådgivning.6 En liten andel av de med en imprintingdefekt har, som tidigare nämnts, en mikrodeletion i imprintingcentret, risken kan då vara upptill 50%.2 Imprinting defekter bör därför leda till vidare rådgivning och testning av familjen.7 Maternell UPD är en de novo sjukdom och risken är mindre än 1%, förutom i väldigt ovanliga fall där UPD uppkommit efter en Robertsonsk translokation. En del data visar på att risken fortfarande är under 1%, men teoretiskt är den högre. Då kan en kromosomanalys vara indikerad. 7,14 Diagnostik Genetiken för Prader Willi är komplex och det kan behövas mer än ett test för att ställa diagnosen. Barnet lämnar ett blodprov och ibland även föräldrarna. Vilken metod som används beror på flera olika faktorer bla om någon analys gjorts tidigare och hur starka misstankarna är för syndromet.6 Men den metod som vanligtvis används för att ställa diagnosen PWS är metyleringsanalys med southern blot.2 DNA metylyserings test med southern blot är ett test som med 99% säkerhet kan ställa diagnosen PWS.7 Detta molekylära test baseras på skillnader i metylering mellan den kvinnliga och manliga homologen. Två olika prober, SNRPN och PW71B, används.15 Normala friska individer har i PWSregionen en metylerad och inaktiv allel från modern och en ometylerad och aktiv allel från fadern. Personer med PWS har i sin PWSregion endast en metylerad allel från modern och analysen visar enbart moderns mönster; då är diagnosen PWS ställd. På detta sätt går det att skilja den friska PWS regionen från den sjuka.15 Metoden kan diagnostisera PWS oavsett vilken kromosomavvikelse som föreligger och kan dessutom differentiera det från AS. 6,2 Dock kan testet inte bestämma vilken molekylär klass som föreligger.6 För att skilja mellan deletion och UPD måste FISH eller PCR utföras.7 En vanlig kromosomanalys utförs vanligen för att detektera andra eventuella abnormaliteter, translokationer och kromosomrearrangemang.7 Den vanliga kromosomanalysen kan detektera endast stora deletioner och andra kromosomavvikelser såsom translokation och extra kromosomer. Dock används inte denna analys metod lika frekvent idag. Vid en deletionsmisstanke bör FISH‐ metoden användas.7 FISH analysen(fluorescence in situ hybridization) kan detektera all typer av deletioner, men den kan inte skilja de två olika deletionerna åt.14 För att finna PWSregionen användes speciella prober i detta fall en SNRPN prob.14 Dock är FISHanalysen bristande när det gäller att diagnostisera för UPD, imprintingdefekter och det går heller inte att avgöra om kromosom 15 kommer från modern eller fadern. Då bör molekylära tester användas. Ibland kan en deletion vara orsakad av en translokation och då bör även en kromosomanalys göras.2 Då diagnosen UPD skall ställas används PCR­baserade sk DNA polymorfismstudier. Blodprov från båda föräldrar och probanden behövs för bästa diagnos. Denna analysmetod används för att avgöra om det är en normal biparental nedärvning eller som i PWS en uniparental nedärvning.7 Metoden kan också säkerställa vissa deletioner.17 Denna analysmetod är inte lika vanlig världen över.6 En imprintingdefekt kan inte diagnostiseras på ett vanligt laboratorium. Oftast missänks imprintingdefekt om en metyleringsanalys är positiv, men andra analysmetoder som gjorts varken talar för UPD eller en deletion. Då kan vidare utredningen på specifika laboratorier med sk sekvensanalys göras. Det är viktigt att fastställa vilken sorts imprinting defekt som föreligger då den ärftliga risken är olika.14 Vilka test som utförs beror givetvis på om något test gjorts tidigare, vilka analys metoder som är tillgängliga och om båda föräldrarna finns tillgängliga för blodprover osv.2 Diagnostiska kriterier Överordnade kriterier: muskelsvaghet, uppfödningssvårigheter, dåligt utvecklad pubertet, dåligt utvecklade könsorgan, fetma, sen psykomotorisk utveckling, omättlig aptit och ett speciellt utseende. Underordnande kriterier: nedsatta fosterrörelser, beteendestörning, sömnsvårigheter, kortvuxenhet, små händer och fötter, ögonproblem, talsvårigheter.4 I USA finns även kriterierna: hypopigmentering, tjockt visköst saliv, artikulerings defekt och skin picking.6 Stödjande fynd: tidig hudpubertet, högsmärttröskel, oförmåga att kräkas, rubbad kroppstemperatur och skolios.4 Prenatal diagnostik PWS kan ej diagnostiseras kliniskt innan födelse.2 Idag används FISH, DNA polymorfismanalys för UPD och DNA metyleringsanalyser för prenatal‐ diagnostik. De flesta laboratorier utför FISH eller PCR.14 Dessa analys metoder kan göras på både amniocentes och CVS.2 PWS är som nämnts tidigare oftast en de novodefekt. Eftersom diagnosen inte kan ställas kliniskt måste något föranleda prenatal diagnostik, om nu inte redan ett barn med PWS redan finns i familjen. Det kan vara så att en kromosomanalys gjorts pga en kvinnas ålder och en abnormalitet har hittats. Om ett barn har PWS pga en deletion eller en UPD är risken för nästkommande barn mindre än 1%. Dessa föräldrar erbjuds rutinmässigt inte prenatal diagnostik. Dock kan dessa föräldrar, om starka önskemål finns, erbjudas prenatal diagnostik för att försäkra sig om att fostret inte har PWS.14 Prenataldiagnostik skall erbjudas de föräldrar där en ärftlig translokation, som ger en deletion, föreligger. Risken är nämligen i dessa fall upp till 25%. FISH kan utföras.14 Om en de novo translokation, som involverar kromosom 15, är funnen bör en FISH och PCR analys utföras.(Detta för att utesluta deletion och UPD)14 Om en familj sedan tidigare har ett barn med PWS pga imprintingdefekt och defekten visar sig sitta i imprinting centrat kan risken var upp till ca 50% att få ännu ett barn till med PWS.18 En DNA metyliseringsanalys skall då erbjudas dessa föräldrar.14 För de familjer med låg risk, dvs utan en familjehistoria med PWS, kan vissa fall leda till prenataldiagnostik. Om en deletion på kromosom 15 misstänks efter någon form av cytogenetiska studier är en FISH‐analys indikerad. Om FISH konfirmerar en deletion bör en analys göras för att identifiera vilken av AS och PWS som föreligger.14 Om en trisomi 15 mosaicism detekteras, bör en PCR eller metyliseringsanalys utföras för att finna ev UPD 15. Trisomi kan finnas i fostret till en början innan en kromosom 15, den från fadern, går förlorad.6,14 Behandling Prader Willi innefattar en rad olika symptom och problem. Det krävs därför insatser från olika yrkeskategorier tex i form av logoped, dietist, arbetsterapeut, läkare, sjukgymnast, psykolog och specialpedagog. Det är viktigt med information om sjudomen till anhöriga och till personen ifråga. Tidigt insatt hjälp med matproblematiken är viktigt. Energiinnehållet måste reduceras till omkring 50‐60% av det normala. Det är viktigt med diet och fasta rutiner kring matintaget. Alltför hård bantning är inte av godo då längdtillväxten kan hämmas. Fysisk aktivitet är också oerhört viktigt. Språkstimulans och teckenkommunikation är till god hjälp innan barnet utvecklat sitt tal. Tandhygien är viktigt då karies drabbar dessa barn i högre frekvens. En defekt i salivens sammansättnig i kombination med ett ökat matintag tros vara orsaken. Barnet kan behöva bettskena då barnen kan ha en annan munhåleanatomi och kan ha problem med sömnapné. Psykologisk testning och skolplacering är viktig för barnets fortsatta utveckling och lärande. De flesta barn går i särskola, men en del går i vanlig klass.1 Tillväxthormontillägg har visat sig vara bra både för längdtillväxt, fetma‐ reduktion och för att muskelmassan skall öka. Även kroppsform och ansiktsform förändras åt det mer normala vid behandling och andningsfunktionen blev bättre . Många PWS‐barn är underventilerade, vilket kan beror på att andningen påverkas av någon signal substans från hypothalamus och denna stimuleras normal från koldioxidhalten i luften. Denna känslighet verkar vara rubbad hos dessa barn, men blir alltså bättre vid behandling med könshormon. Har barnen en brist bör en behandling sättas in så snart som möjligt så att barnet inte tappar i längdtillväxt. Behandlingen kräver att PWS är verifierad och att barnet står på diet. Det har visat att utan diet är effekten av behandling ej lika god. Sedan 2000 är det accepterat att behandla barn med tillväxtrubbning med tillväxthormon, efter att det tidigare varit ett hårt motstånd.4, 10 Behandling med könshormon bör övervägas när slutlängd uppnåtts och då personen inte har någon pubertet. Kontroll av gonadotropiner och könshormon bör utföras.1,11 Testosteron ges till en del män i 20 årsåldern, de får då bättre muskelmassa och bättre ämnesomsättning. Könshormonsbehandling till kvinnor är mer restriktiv, då det är osäkert om flickor behöver substitution av könshormon eller om det är tillräckligt med östrogenproduktion från den ökade fettmängden.9 Det är också viktigt att uppmärksamma beteendestörningar hos PWS. De är ofta svåra att åtgärda. Det finns ingen bestämd behandlingsåtgärd för beteende störningar eller problem. Men det är viktigt att identifiera dessa problem tidigt för att kunna gå in med hjälp och stödinsatser. Självdestruktiva beteende kan behöva individuell behandling och finns ångest eller tvångsbeteende kan psykofarmaka komma att behövas.1 SSRI tros kunna öka mättnadskänslan och kan vara till hjälp för dessa barn. Ibland kan antipsykosmedel mot vredesutbrott behövas, men det ger dock mkt biverkningar.4 Funderingar kring etiska frågeställningar En viktig fråga att diskutera är den prenatala diagnostiken. Idag används prenatal diagnostik för att upptäcka kromosomavvikelser. Jag har förståelse för att de föräldrar, som sedan tidigare har ett barn med PWS, vill ha en möjlighet att göra prenatal diagnostik. Det är mer belastande för en familj där ett barn inte är lika välfungerande som normala barn. Risken med prenatal diagnostik är dock att vi i dagens samhälle väljer att endast de barn utan några som helst svagheter är värdiga ett liv. Det viktiga är att vår kunskap kring syndromet utvecklas så att vi kan lära och utforma hjälpmedel och stöd, som ger ett värdigt liv för dessa barn och deras familjer. Det är också viktigt med genetisk rådgivning till dessa föräldrar. Inte enbart för att visa vilken typ av kromosomavvikelse som föreligger, utan kanske snarare att hjälpa och stötta familjerna. Man ska inte utgå från att föräldrarna vill avbryta graviditeten om man får besked om en kromosomavvikelse tex PWS, utan lyssna och följa familjens funderingar och önskemål. Jag tror det är viktigt med information kring syndromet, men även information om det stöd, den hjälp och de behandlingsinsatser som finns för dessa barn. För att vi ska förstå mer om PWS är forskning givetvis viktigt. Detta för att förstå vilka gener som är påverkade vid syndromet. Men det är också viktigt att satsa på medicinska, psykologiska och pedagogiska insatser för att ge hjälp och stöd åt barnen, föräldrarna och syskonen. Allt detta för att ge möjligheter till en hög livskvalitét för barn med PWS. Sammanfattning Varje år föds 6‐8 barn med PWS. Syndromet beror på en kromosomavvikelse på den långa armen av kromosom 15. De olika mekanismerna, deletion, UPD och imprinting, leder alla till undertryck av gener i en viss region av faderns kromosom 15. Flera gener är intressanta i PWS regionen men forskare vet än idag lite om vilken/vilka gener som är ansvariga. Huvudsymptomen är en oförmåga till mättnadskänsla, fetma, utvecklingsstörning, hypogonadism och muskelhypotoni. Ärftligheten är mindre än 1% för de flesta avvikelserna. Dock finns en högre risk vid en imprintingdefekt eller en translokation med en deletion. Diagnostiken för PWS görs mha kromosomanalys, FISH, PCR och Southern blot. PWS är ett komplext syndrom och dessa barn behöver hjälp från flera olika yrkeskategorier. Det är viktigt att skapa ett värdigt och gott liv för dessa barn. Referenslista 1)Socialstyrelsens hemsida http://www.socialstyrelsen.se/ovanligadiagnoser/Prader‐Willis+syndrom 2) European journal of human genetics(2009) ­Prader Willi syndrome http://www.nature.com/ejhg/journal/v17/n1/full/ejhg2008165a.html 3) Emerys , Thompson and Thompson­Genetics in Medicine sixth edition. 4)Ågrenska 2001 http://www.agrenska.se/Global/Nyhetsbrev/PWS%202001.pdf 5) Prader­Willi syndrome: intellectual abilities and behavioural features by genetic subtype http://www3.interscience.wiley.com/journal/118735326/abstract 6)The genetics of Prader Willi Syndrome http://www.pwsausa.org/syndrome/Genetics_of_PWS.htm 7) Diagnostic testing for Prader ­Willi and Angelman syndromes. http://genetics.faseb.org/genetics/acmg/pol‐22.htm 8)Mortality in Prader Willi Syndrome http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2422866 9)Behandling av Prader Willi Syndrom http://www.tillvaxthormon.se/default____12782.aspx 10) Tillväxthormon behandling av barn med Prader ­Willi Syndrom http://www.lilly.no/Nitro2/objects/pdf/veksthormon/no_pedendo_praderwilli_ volum12.2_98.pdf 11)Riktlinjer för omhändertagande av individer med Prader Willi syndrom. http://www.blf.net/endodiab/vardprogram‐pws.doc 12)Karolinska institutet http://www.prader‐willi.se/martinritzenPWS07.ppt#3‐svensk 13)Gener och individuella skillnader i beteende http://www.lakartidningen.se/07engine.php?articleId=9644 14)Prader Willi Syndrome http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=pws 15)Management of Prader Willi Syndrome http://books.google.se/books?id=HnVOj5_rLpIC&pg=PA79&lpg=PA79&dq=PW7 1B&source=bl&ots=19om6n78j8&sig=NTa0xcjXvFASXwSkFCwYPoB6vCo&hl=sv &sa=X&oi=book_result&resnum=1&ct=result#PPA79,M1 16) Behavioral Differences Among Subjects With Prader­Willi Syndrome and Type I or Type II Deletion and Maternal Disomy http://pediatrics.aappublications.org/cgi/content/full/113/3/565 17)Prader­ Willi syndrome http://www.allkids.org/body.cfm?id=142