Genetisk forskning – nyckeln till framtidens behandlingar
advertisement

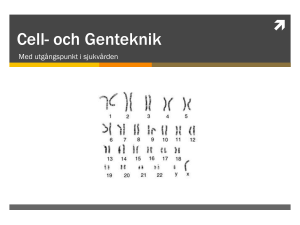
Genetisk forskning – nyckeln till framtidens behandlingar Irina Golovleva Sjukhusgenetiker Inst. för medicinsk biovetenskap, klinisk genetik Många gener är involverade i ögonsjukdomar, men med tanke på hur stor hela arvsmassan är talar vi ändå om en mycket liten del. Arvsmassan hos människan finns i form av DNA som innehåller all genetisk information. DNA är inpackat i kromosomer (figur 1) och det finns exakt 23 par kromosomer: 22 par s.k. autosomala kromosomer och ett par könskromosomer som skiljer män från kvinnor, XX hos kvinna och XY hos man. Kromosomerna ligger i cellkärnan i kroppens celler, se figur 1. Figur 1. Arvsmassans packning och plats i människokroppen. 27 DNA består av fyra byggstenar, nukleotider: adenin, tymin, cytosin och guanin. Förr uppskattade man att det finns mellan 50 000 och 100 000 gener men numera, när nästan hela människans genom är känt, vet man att det korrekta antalet ligger mellan 20 000 och 30 000 gener. Av detta är inte de 120 eller 200 gener som är inblandade i ögonsjukdomar en särskilt stor andel. Dessutom är bara 30 procent av innehållet i hela genomet gener, medan 70 procent är det man förr kallade ”junk-DNA” (”sop-DNA”). Men det är inte riktigt sant, för där finns viktiga DNA-delar som reglerar olika proteiners funktion. Dessutom finns åtskilliga sekvenser som man ännu inte vet funktionen för. Om man tittar på genernas struktur är exoner (delar av gener) viktiga för att senare bygga upp protein. Regleringssekvenser ligger framför genen medan intronsekvenser är DNA mellan olika exoner, se figur 2. Genom en process som heter klyvning, splicing på engelska, tas intronerna bort. Därefter byggs ”budbärar-RNA”, mRNA, som översätts till proteiner. Figur 2. Genstruktur i arvsmassan. Mellan ”start” och ”stop” i figuren ligger en gen som efter klyvning kan koda för ett bestämt protein. DNA är alltså uppbyggt av fyra nukleotider. Tre nukleotider på rad (en triplett) bygger upp en kod, en ”bokstav”. Varje triplett översätts till en aminosyra. Det finns därmed ganska många DNA-variationer, och det skulle vara konstigt om det inte vore så eftersom människor är olika pga. variationer i DNA. Det finns olika typer av variationer, både ”bra” variationer och ”dåli28 ga” variationer. Med ”bra” i det faller menas att när en nukleotid byts ut mot en annan blir i slutänden båda proteinerna fungerande. Utbytet påverkar inte funktionen och det är ingen fara för människan. ”Dåliga” utbyten däremot resulterar i ett icke-fungerande protein. Det kallas mutationer och associeras framför allt med olika sjukdomar. Som ett enkelt exempel kan vi som i figur 3 tänka oss att varje ord är en aminosyra som alltså består av tre nukleotider. I normalfallet ska DNA-koden översättas till proteinet ”grå mus ser oss med ett öga”. Vid en mutation av typen punktmutation, där en nukleotid byts mot en annan, t.ex. om s i ordet oss byts mot t, innebär missens att man får proteinet ”grå mus ser ost med ett öga”. En annan mutationstyp kallas nonsens och betyder att hela proteinet blir kortare och inte fungerar som det ska, t.ex. ”grå mus ser”. En typ som kallas deletion innebär att några nukleotider (bokstäver) kan vara borta, t.ex. ”grå mus med ett öga”. Slutligen finns mutationstypen insertion, som innebär att ytterligare en nukleotid kommer in i DNA-sekvensen och då tappar meningen/proteinet sin betydelse. Figur 3. Förändringar i arvsmassan som ger icke-fungerande protein kallas mutationer. Här åskådliggörs de mutationstyper som man känner till. Dessa typer av mutationer förekommer vid olika sjukdomar. Jag tänkte ge exempel på två som orsakas av punktmutationer. Det är viktigt att veta hur mutationerna vid olika sjukdomar ärvs. Här koncentrerar vi oss på de två typer som kallas autosomalt dominant och autosomalt recessiv nedärvning. Or29 det ”autosomal” betyder att sjukdomsgenen ligger på en av de 22 autosomala kromosomerna och inte på en könskromosom. Om vi börjar med autosomalt recessiv nedärvning, visar figur 4. det som kallas släkttavla för de tidigare generationer som man har information om. Fyrkanterna står för män och ellipserna för kvinnor; de mörkare är friska individer och de ljusare är sjuka. I figuren är fyra generationer helt friska och i femte generationen kommer två sjuka. Man kan också se att de sjukas föräldrar är kusiner, vilket ökar risken att få sjukdomen. Vid recessiv nedärvning är föräldrarna alltid friska men bärare av mutationen. Deras risk för att få ett sjukt barn är 25 procent. Figur 4. Släkttavla med autosomalt recessiv nedärvning. Fyrkant = man, ellips = kvinna, mörk = frisk, ljus = sjuk. Nedärvningen av botniadystrofi Ögonsjukdomen Botniadystrofi är en typiskt autosomalt recessiv sjukdom. På släktkartan i figur 5 finns friska föräldrar med sjuka barn, som i Figur 4. Genen för botniadystrofi finns på kromosom 15 och översätts till proteinet CRALBP. Den punktmutation som ändrade DNA-koden gav som resultat ett utbyte av aminosyran i position 233. Redan innan man visste att det proteinet är associerat till ögonsjukdomar visste man dess funktion: CRALBP är ett viktigt protein i rodopsinomsättningen (syncykeln) och fungerar som bärare av vitamin A. 30 Figur 5. Nedärvningskarta över ögonsjukdomen botniadystrofi, en autosomalt recessivt nedärvd sjukdom. Även här får friska föräldrar sjuka barn (mörka markeringar), jfr Figur 4. Figur 6 visar RLBP1-genen, som orsakar botniadystrofi. Den består av åtta exoner och man har hittat totalt nio mutationer. Andra forskargrupper hittade enstaka, maximalt fem, sjuka individer, men vi hittade ca 75 patienter med just den mutation vi studerar. Vi lyckades också utveckla en ganska enkel metod för genetisk testning, där man från blodprov kan se om en person bär på anlaget. Vid recessivt ärvd sjukdom måste patienten bära på dubbla anlag för att sjukdom ska uppstå. Vi kan nu erbjuda genetisk testning och vägledning där vi bedömer risken för att familjen får barn med samma sjukdom. Däremot kan vi inte göra någonting åt sjukdomen som sådan eftersom det inte finns något botemedel. Vi studerade också hur mutationen påverkar proteinet och kunde påvisa att protein som bär på mutationen R233W binder till vitamin A betydligt starkare än normalt protein. Det kan vara denna mekanism som orsakar botniadystrofi. Vi har också studerat alla patienter från norra Sverige med retinitis pigmentosa. Bland dem fann vi 65 med dubbel uppsättning av R233-mutationen och 10 med bara enkel uppsättning av mutationen, se figur 6. Därför letade vi efter ytterligare en mutation och lyckades identifiera den förra året. Den ligger inte långt borta från den första, närmare bestämt i position 225. I de familjerna kan man se ett typiskt autosomalt recessivt mönster. Föräldrarna är friska och anlagsbärare, men bär på olika mutationer i samma gen. Deras farfar och mormor bär också på de olika mutationerna. Om farmor och morfar skulle ha gift sig finns risken att de skulle har fått barn med sjukdomen, 31 men det blev inte så utan deras barn fick bara anlaget. Sedan träffades barnen och deras pojke fick båda mutationerna, så den genen ger som resultat ett icke fungerande protein. Figur 6. Genen RLBP1, som orsakar ögonsjukdomen botniadystrofi, består av åtta exoner och man har hittat totalt nio mutationer i denna gen. Umeåforskarna har intresserat sig för mutationen vid nr 7 i figuren och utvecklat en testmetod för den. Den nedre bilden återger testresultat från blodprov: Anlagsbärare visar tre band, en frisk människa har två band och en sjuk bara ett band. Man kan förstås fråga sig varför det finns så många fall av botniadystrofi i vår region. Det finns en individ i Japan, en familj i Kina, men här har vi 75 personer. Det finns i princip ingen slump bakom detta utan det beror på geografi, kultur och sociala aspekter. En viktig faktor är den s.k. founder-effekten, se figur 7, som kan förklara en stor del av de genetiska sjukdomarna i norra Sverige. 32 Figur 7. Den s.k. founder-effekten är en viktig del av förklaringen till att ärftliga sjukdomar koncentreras till vissa, glest befolkade områden. En människa som bär på anlaget kommer in i en befolkning och lever där. Vid isolering av området, t.ex. krig eller folkomflyttning, minskar befolkningen medan frekvensen av anlagsbärare ökar. När befolkningen sedan växer på nytt ökar risken för att mutationen sprids och andelen anlagsbärare ökar. Genterapi en spännande möjlighet När man har en så stor grupp av patienter som vi undrar man desto mera kring möjligheterna att bota sjukdomen. Genterapi är förstås ett jättespännande alternativ – att ersätta en icke-normal, muterad gen med en normal ickemuterad är då målet. Men innan man kommer fram till att kunna testa något sådant måste man noga studera vilken gen som orsakar sjukdomen, vilka mutationer som finns i genen, vilken roll varje mutation spelar för proteinets funktion och vilka celltyper som påverkas av ett icke-fungerande protein. De som ska genomgå genterapi måste tänka på vilka fördelar man kan nå men också vilka risker man måste ta. Den genterapi som levereras genom virus kanske inte fungerar helt rätt. Kanske kan immunsystemet inte ta emot genterapi. Vem är vinnare, vem kan bli förlorare och vad betyder det för patienten, hans eller hennes familj och samhället? Man fortsätter testa genterapi på framför allt möss, men det är långt kvar till motsvarande framsteg på människa. 2001 publicerades en artikel i Nature Genetics om genterapi på stora hundar. Man kände till en sjukdom, Lebers congenital amaurosis, som innebär att hundar föds blinda. Man analyserade genomet och identifierade en recessiv mutation i genen RPE65 som faktiskt interagerar med CRALBP. Därför behandlades hundarna med genterapi, genom virus som bär på en normal kopia av RPE65-genen, och blinda hundar fick faktiskt tillbaka synen. Men man gjorde ett schema, se figur 8, över den långa vägen från forskning till behandling. I USA är kliniska prövningar på människa med Leber congenital amaurosis fortfarande inte igång (2007). 33 Figur 8. Den långa vägen från forskning till fungerande, säker och godkänd behandling. Ett annat sätt på vilket mutationer går i arv är autosomalt dominant nedärvning. Här är en av den sjukes föräldrar alltid bärare av mutationen och här finns en risk på ca 50 procent för att få ett sjukt barn. Den här typen av dominant nedärvning har ögonsjukdomen tappdystrofi, där tapparna är påverkade. Vi samlade DNA från två stora familjer där denna sjukdom förekom. Det finns individer med sjukdomen i varje generation, vilket visar att tappdystrofi har autosomal dominant nedärvning. Med s.k. mappningsanalys och statistisk metod lyckades vi ringa in genen PITPNM3, en ganska stor gen med 20 exoner. Man har förut visat att en liknande gen som finns hos bananflugan, Drosophila, och i muterad form kan orsaka fotoreceptordöd hos flugan. Med sekvensanalys, som visar nukleotidernas ordningsföljd, kunde vi påvisa ett nukleotidutbyte och som resultat också ett aminosyrautbyte i proteinet. Vi visade att 322 friska individer i materialet inte hade den mutationen och var då ganska säkra på att vi funnit den mutation som ger sjukdom hos patienterna. Den fanns hos alla sjuka i släkten. Därmed kunde vi göra ytterligare en genetisk testning på alla patienter i den släkten och gå vidare med att undersöka andra med liknande sjukdom. När vi vet vilken mutation som orsakar sjukdomen vill vi naturligtvist studera proteinet, som kan hittas i hjärna, mjälte och äggstockar. Det påverkar tapparnas funktion. Hos fiskar finns proteinet mest i tapparna och tapparna är också mest påverkade hos tappdystrofipatienter. I vår fortsatta forskning 34 vill vi studera hur det muterade proteinet fungerar och vilka mekanismer som står bakom sjukdomen. Vi vill också göra en modell av sjukdomen på bananfluga, fisk eller mus. Arbetet har gjorts av många studenter som genom åren har passerat klinisk genetik. Det är gjort i samarbete mellan oftalmiatrik och medicinsk/klinisk genetik. Marie Burstedt skrev avhandling om en botniadystrofi, Ola Sandgren kläcker idéer, träffar patienter och samlar blodprover, gästforskaren Konstantin Kadzhaev jobbade också med botniadystrofi medan Linda Köhn identifierade sjukdomsgenen hos patienter med tappdystrofi. Sverker Olofsson: Vi har haft tio Forskningens dag och väldigt ofta kommer man in på det här med gener och då dyker ordet genterapi upp. Det betyder alltså att byta ut en dålig gen mot en bättre? Irina Golovleva: Ja, det är tanken. Men det finns fler möjligheter, t.ex. kan man hämma eller sätta något på den felaktiga genen för att den inte ska fungera längre utan bara den normala som stoppas in. Sverker Olofsson: Det är ju lätt när man följer de här resonemangen att undra var genen egentligen sitter. Var plockar man ut den och sätter in den nya? Hur går det till att byta ut den där skadade genen? Irina Golovleva: En normal gen måste produceras på laboratoriet. Det kan göras från vilken frisk människa som helst som inte bär på en mutation. Sedan måste man också ”leverera” en normal gen på rätt sätt. Om man tar in en gen som endast finns i näthinneepitelet och stoppar in den i någon annan vävnad fungerar det inte alls. Det är jätteviktigt att leverera till rätt celler. Sverker Olofsson: Men hur får man generna, är det med dropp? Sprutar man in dem eller opererar man? Hur fixar man det? Irina Golovleva: Man kan spruta in dem, men då måste man veta att de hamnar rätt. Ett alternativ är att ta bort celler från kroppen, injicera friska gener i dem och sedan stoppa tillbaka dem. Det finns också andra möjligheter, men för det mesta använder man spruta i form av någon lösning. Sverker Olofsson: Du som håller på med det här, ser du en framtid där rätt mycket av hälsovården kommer att handla om att fixa och trixa med generna? 35 Irina Golovleva: Jag vill inte vara skeptisk, men det är i så fall ett jättelångt perspektiv. Alla sjukdomar kan inte botas med genterapi på grund av att vi ändå vet så lite. De bästa kandidaterna är de sjukdomar som orsakas av en enda gen. Sverker Olofsson: Men man kan säga att de här hundarna som blev seende kan alltså vara människor om ett visst antal år? Irina Golovleva: Ja, så kan det bli för i USA är det redan igång. Prekliniska studier är utförda och man förbereder sig för kliniska prövningar. Sverker Olofsson: Fem år, tio år? Irina Golovleva: Jag vill vara optimist och säger fem år, åtminstone för fas ett och två, men sista stadiet med kliniska prövningar ska göras på 3 000 människor. Det blir jättesvårt att hitta en så stor grupp med en ögonsjukdom. 36