Skräp-DNA?? - Nationellt resurscentrum för biologi och bioteknik
advertisement

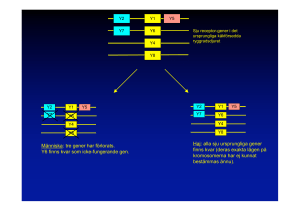
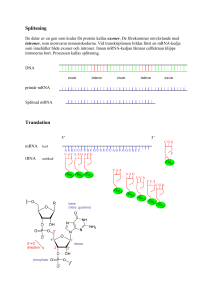
Övning i bioinformatik Nationellt resurscentrum för biologi och bioteknik Skräp-DNA?? Ett av de hetaste vetenskapliga fälten idag är det område där biologi, informationsteknologi och datavetenskap kombineras till en enhet som kallas bioinformatik. Ca 98% av mänskligt DNA kodar inte för proteiner och har ingen känd funktion. Det kallas ibland skräp-DNA (”junk” DNA), men det är egentligen inte något bra ord eftersom skräp associerar till något som inte behövs och som vi kan kasta bort, men det som vi kallar skräp-DNA kanske har funktioner som vi inte förstår idag. I det här sammanhanget definierar vi skräp-DNA som avsnitt av DNA som inte kodar för gener. Skräp-DNA finns både inne i gener (introner) och mellan gener. Själviskt DNA? En del av detta DNA-matererial består av specifika sekvenser av nukleotider som upprepas ett stort antal gånger och som inte verkar ha någon känd funktion. Long interspersed elements (LINES) och short interspersed elements (SINES) är två klasser med sådana repetitiva sekvenser. De är retrotransposoner, hoppande DNA som förökar sig och förflyttar sig inom genomet. Kanske kan man betrakta detta som själviskt DNA som parasiterar på en cell för att själv kunna förökas? L1 (ca 15% av genomet) är exempel på LINES och Alu-sekvenser (ca 10% av genomet) hör till SINES. I denna övning ska du studera relationen mellan “skräp”-DNA och gener, samt uppbyggnaden av gener (promotor, introner, exoner, poly-A). Metod Tre Internetsidor används i den här övningen för att studera gener från olika organismer: • National Center for Biotechnology Information (NCBI), www.ncbi.nlm.nih.gov • The Sequence Manipulation Suite, www.ualberta.ca/~stothard/javascript/ • GENSCAN, http://genes.mit.edu/GENSCAN.html 1. Som exempel kan följande gener studeras: • Xenopus Cad2 gene, NM_204085 • Chicken H1 histone gene, M17019 • Zea Mays alcohol dehydrogenase, M32984 • Norway Rat neurogranin RC3 gene, U22062 • Arabidopsis thaliana DNA chromosome 4 fragment, Z97343 (En alternativ möjlighet att välja gener att undersöka beskrivs nedan.) 2. Gå in på webbsidan från NCBI (www.ncbi.nlm.nih.gov). 3. T.v. i övre delen av sidan står Search. Välj Nucleotide från menyn omedelbart till höger. (Detta innebär att det blir möjligt att söka efter nukleotidsekvenser i NCBI:s databas.) 4. Välj en gen att studera (t.ex. en av ovanstående gener). Skriv in beteckningen för genen (t.ex. M32984) i rutan t.h. Klicka på GO. (Om namnen på generna ovan skrivs in i stället får man för vissa av generna (t.ex. ”Zea Mays alcohol dehydrogenase”) en alltför stor mängd träffar.) 5. En ny sida öppnas med en klickbar länk. Länken leder i sin tur till en ny sida med en länk till en sida som innehåller en mängd information, bl.a. finns DNA-sekvensen för genen längst ner på sidan. Nationellt resurscentrum för biologi och bioteknik • Bioinformatikövning 2005.11.08 • Får fritt kopieras i icke-kommersiellt syfte om källan anges 6. Markera och kopiera (Ctrl + C) DNA-sekvensen längst ner på sidan. 4. Gå in på The Sequence Manipulation Suite (www.ualberta.ca/~stothard/javascript/). Klicka på länken Filter DNA som finns t.v. högt upp på sidan. 5. På den sida som öppnas finns en textruta som eventuellt redan innehåller en DNA-sekvens. Klicka på Clear för att ta bort denna. 6. Klistra in din egen sekvens (Ctrl+V) i rutan. Tryck på Submit för att ta bort siffrorna som står först i varje rad med nukleotider. (Det går inte att söka på nukleotidsekvensen innan dessa siffror tagits bort.) 7. Markera (Ctrl + A) och kopiera den redigerade sekvensen (Ctrl+C). 8. Öppna sidan GENSCAN (http://genes.mit.edu/GENSCAN.html). 9. Klistra in den kopierade nukleotidsekvensen i den stora tomma textrutan en bit ner på sidan. 10.Välj organismtyp (Vertebrate, Arabidopsis, Maize) i menyn ovan textrutan. 11.Klicka sedan på Run GENSCAN (står under rutan med den inklistrade DNA-sekvensen). 12.Resultatet innebär en mängd information om genen, exoner, promotorregion, poly A svans och den förmodade aminosyrasekvensen. (Se exempel M32984 (majs) med förklaringar på nästa sida.) 13. Beräkna andelen nukleotider (baspar) i genen i förhållande till det totala antalet nukleoti­der för sekvensen genom att ta reda på hur många baspar hela sekvensen består av, samt hur många aminosyror som ingår i det protein som sannolikt kommer att bildas. (Totala antalet baspar står i övre delen av webbsidan och totala antalet aminosyror (enbokstavskod) står omedelbart ovanför aminosyrasekvensen.) Eftersom en aminosyra kodas av tre nukleotider multipliceras antalet aminosyror med tre. Antalet kodande nukleotider divideras sedan med totala antalet nukleotider i sekvensen. Detta ger andelen av den kodande regionen i för hållande till hela sekvensen. 13.Klicka på Click here to view PDF image of the predicted gene(s). Du får då fram ett överskådligt diagram som visar exoner, introner och i vilken riktning generna läses av. 14.Studera M32984 från majs a) Hur många gener finns totalt? b) Var börjar och slutar genen? c) Hur många introner och exoner innehåller den? 15. Studera på samma sätt Z97343 från Arabidopsis thaliana. Hur många gener finns totalt i vardera avläsningsriktningen? Alternativ möjlighet att välja och studera gener 1. Gå in på webbsidan från NCBI (www.ncbi.nlm.nih.gov). 2. Till höger finns rubriken Hot spots och en bit ner finns länken Human genome resources. 3. Länken leder till en ny sida där det t.v. finns figurer som schematiskt visar människans kromosomer. Klicka på en av kromosomerna. 4. På den sida som öppnas finns en karta över denna kromosom där de kända generna anges. Härifrån kan beteckningar på gener hämtas och användas för att studera gener i den föregående övningen. Vissa av generna har t.h. en länk ”OMIM”. Denna länk ger en beskrivning av genens funktioner. 5. Om du klickar på någon av generna får du fram information om genen vilken inkluderar en karta över genen som visar kodande och ej kodande regioner. (Genkartan är inte lika detaljerad som den karta du får fram i övningen ovan. Referens Övningen bygger på och är ett sammandrag av nedanstående artikel: Visualising ”junk” DNA through bioinformatics Nancy L Elwess, Sandra M Latourelle, Olivia Cauthorn, Plattsburgh State University, USA. Journal of Biological Education (2005)39(2) Artikeln kan hämtas från: www.iob.org/downloads/236.pdf Nationellt resurscentrum för biologi och bioteknik • Bioinformatikövning 2005.11.08 • Får fritt kopieras i icke-kommersiellt syfte om källan anges Nedan visas resultatet för genen från majs (M32984) efter GENSCAN. (Det blir små variationer från gång till gång.) Endast det som är speciellt intressant att lyfta fram har tagits med, övrig information har tagits bort (i tabellen finns t.ex. endast några av kolumnerna med). Kommentarer/förklaringar har skrivits in i texten nedan. GENSCANW output for sequence 04:15:10 GENSCAN 1.0 Date run: 14-Oct-105 Time: 04:15:10 Sequence 04:15:10 : 6225 bp : 43.95% C+G : Isochore 1 ( 0 - 100 C+G%) Parameter matrix: Maize.smat Predicted genes/exons(Förklaring till kolumnerna i tabellen, se nedan): Gn.Ex Type S .Begin ...End .Len ----- ---- - ------ ------ ---1.00 Prom 557 518 40 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 Prom Init Intr Intr Intr Intr Intr Intr Term PlyA + + + + + + + + + + 1129 1275 1843 2077 2540 3545 3694 3881 4143 4300 1168 1308 1979 2123 2865 3606 3789 4042 4259 4305 40 34 137 47 326 62 96 162 117 6 3.00 Prom + 3.01 Init + 5057 5370 5096 5414 40 45 Click here to view a PDF image of the predicted gene(s). PDF-bilden ger en mycket bra översikt över genen med de olika komponenterna som ingår, se figur nedan. Predicted peptide sequence(s)(Nedan visas den förmodade aminosyrasekvensen): >04:15:10|GENSCAN_predicted_peptide_2|326_aa MATAGKVIKCKAAVAWEAGKPLSIEEVEVAPPQAMEVRVKILFTSLCHTDVDFWEAKGQT PVFPRIFGHEAGGIIESVGEGVTDVAPGDHVLPVFTGECKECAHCKSAESNMCDLLRINT DRGVMIADGKSRFSINGKPIYHFVGTSTFSEYTVMHVGCVAKINPQAPLDKVCVLSCGIS TARKFGCTEFVNPKDHNKPVQEVLAEMTNGGVDRSVECTGNINAMIQAFECVHDGWGVAV LVGVPHKDAEFKTHPMNFLNERTLKGTFFGNYKPRTDLPNVVELYMKKELEVEKFITHSV PFAEINKAFDLMAKGEGIRCIIRMEN >04:15:10|GENSCAN_predicted_peptide_3|15_aa MENDKGVFWKVSFPR Explanation(till tabellen ovan): Gn.Ex : gene number, exon number (for reference) Type : Init = Initial exon (ATG to 5’ splice site)(Den första exonen i genen.) Intr = Internal exon (3’ splice site to 5’ splice site)(Exoner inne i genen.) Term = Terminal exon (3’ splice site to stop codon)(Avslutande exon.) Sngl = Single-exon gene (ATG to stop)(Genen består av endast en exon.) Prom = Promoter (TATA box / initation site)(Promotor, relgerar genuttrycket.) PlyA = poly-A signal (consensus: AATAAA) S : DNA strand (+ = input strand; - = opposite strand) Begin : beginning of exon or signal (numbered on input strand) End : end point of exon or signal (numbered on input strand) Len : length of exon or signal (bp = antal baspar) Nationellt resurscentrum för biologi och bioteknik • Bioinformatikövning 2005.11.08 • Får fritt kopieras i icke-kommersiellt syfte om källan anges Figuren nedan visar sekvensen M32984 från majs och uppdelningen i introner och exoner. Figuren nedan visar en del av sekvensen Z97343 från Arabidopsis. Här syns ett betydligt mer komplicerat mönster av olika gener. Ovan linjen (DNA-molekylen) med markeringar som visar kb (tusenbaspar) syns sju gener med introner och exoner. Nedanför linjen visas fem hela gener och två delar av gener längst t.v. och t.h. Generna ovan respektive under linjen (DNA-molekylen) läses av motsatta riktningar som visas av pilarna Nationellt resurscentrum för biologi och bioteknik • Bioinformatikövning 2005.11.08 • Får fritt kopieras i icke-kommersiellt syfte om källan anges